Abstract
Enzymes retain their activity and stability over a range of temperatures and time. Typically, that is the case with Neutrase, which is obtained from Bacillus Amyloliquefacians. Experimental data showed behavioral changes for Neutrase when subjected to temperature and time parameters. It showed a denaturing effect of temperature and time on the stability of the enzyme. Typically, experimental data showed significant changes in the activity of the enzyme, with a positive gradient when a graph of absorbance against temperature change was plotted. Other values used to plot the graph of time against the absorbance rate showed a strong relationship with time and enzyme stability.
Typically, a positive gradient was obtained showing that enzyme activity could rise with time until a state was attained where the activity was static. To explain the enzyme behavior when subjected to thermal and time variables, theoretical propositions underpin the rationale for the observations. Typically, the thermal stability seems to be associated with the structure of the molecules and their exposure to the reacting surface. That is attained by the modification of the molecular structure of the Neutrase enzyme with rising in temperature, thus, suggesting the use of additives to provide an additional increase in the lifespan of the catalyst.
Introduction
According to Fukumoto (1943), neutralize is a hyperthermophilic protease enzyme derived from the Bacillus amyloliquefaciens bacterium. Neutrase belongs to the family of proteolytic enzymes, which are present in all forms of living organisms. A critical study and analysis of the family of proteolytic enzymes show that they belong to a distinct family of proteins. Typically, proteolytic enzymes have a unique property that allows them to become active when introduced to the appropriate chemical environment and become inactive, existing as inactive zymogens when produced. These findings were established by comparing the molecular sequence of amino acids, which are three-dimensional structures, with the protease enzyme reaction mechanisms. The distinguishing factor is the relationship and underlying behavior due to the molecular sequence of the protease and its reaction mechanisms.
Experimental results show that there are differences in molecular patterns that reveal significant differences between proteolytic enzymes and other enzymes but reveal structural patterns for each enzyme that are similar to a specific family of proteins. The differences and similarities are found in the molecular structures of the enzymes. However, it is crucial to note that their molecular structures and resulting chemical behavior have a strong bearing on their thermal stability when subjected to thermal denaturation and storage time.
Protease enzymes are used to hydrolyze the link between protein molecules due to peptide bonds. Typically, Protease enzymes take other names such as proteolytic enzymes, with varying catalytic activities. When carrying out the catalytic activities, as already mentioned, peptide bonds are broken by the intervention of proteolytic enzymes. The bonds between the molecules are covalent bonds formed through a condensation reaction that involves the release of H2O molecules. The condensation reaction that releases the H2O molecules occurs between an amino group of molecules and a carboxyl group (Knapp, de Vos, Rice & Ladenstein, 1997).
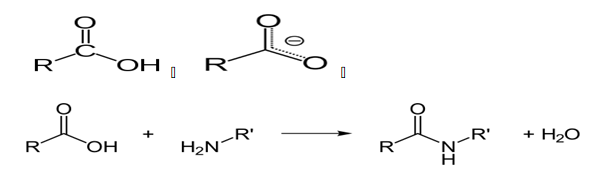
The reaction between the carboxyl group and the peptide group to give an amide belonging to the peptide family and a water molecule is shown above. It is crucial to note the peptide family of chemicals is large, with extensions such as esters, with varying thermal and decomposition properties (Klingeberg, Galunsky, Sjøholm, Kasche & Antranikian, G.1995).
Proteolytic enzymes are known to have evolved with changes occurring in their molecular structures, and underlying functions. That has caused changes in their inhibiting characteristics, with their inhibitors functioning differently. Typically, the inhibitors play simple roles in manipulating activities in primitive organisms while taking more complex roles in higher organisms.
Studies show that proteolytic enzymes perform physiological functions ranging from overall protein digestion to more specific processes that are regulated. Examples of regulated processes include the activation of zymogens, blood coagulation and lysis of fibrin clots, the release of hormones and pharmacologically active peptides from precursor proteins, and transporting secretory proteins across membranes (Kirino, Aoki, Aoshima, Hayashi, Ohba, Yamagishi, Wakagi & Oshima, 1994).
The overall application of neutrase has been applied in the food and agricultural industries. Typical examples include studies by Skeie (1995) who showed that adding Neutrase to low-fat gouda-type cheese could increase proteolysis after the first-day cheese was made. The resulting cheese containing neutrase developed higher levels of amino N substances and acetaldehyde along with other volatile compounds. It was demonstrated that by adding neutrase to cheese, the cheese texture showed significant improvements by developing a firmer texture (Kimura, Kanaya & Nakamura, 1992).
According to Ovissipour et al (2009), neutrase is an endo-protease substance that is used in most cases in reactions that require proteins to be broken down either moderately or extensively into peptides. Thus, the underlying properties of neutrase make it an appropriate chemical for use in the food and agricultural industry mainly in the fermentation of proteins, whose origin is in animals or vegetables.
On the other hand, neutrase has shown a significant impact in discoveries when applied to investigate enzymatic hydrolysis processes on proteins derived from animals e.g. Fish. The study, as reported by Ovissipour et al (2009) shows that microbial enzymes such as Neutrase offer several advantages, which include significant catalytic activities and are stable at higher pH concentrations and temperatures (Ovissipour et al 2009). One of the properties identified with normal Neutrase is that it contains the neutral strain of Bacillus amyloliquefaciens proteases only though it can be modified commercially to contain alkaline proteases, as an additional property (Kim, Kim, Bae, Yu & Oh, 1998).
According to Sigma (2004), the process of creating enzymes with different activities is based on different synthesizing methods. Accordingly “Enzymes synthesized by hyperthermophiles (bacteria and archaea at optimal growth temperatures of >80°C), also referred to as hyperthermophilic enzymes, are typically thermostable (i.e., resistant to irreversible inactivation at high temperatures)” (Sigma, 2004). That shows that “optimally high reaction activities occur at higher temperatures than at lower temperatures” (Sigma, 2004). Inactivation of enzymes is based on the equilibrium shift of the enzyme with thermal stability that is under investigation.
However, such enzymes can only be extracted at safe temperatures lower than 1150C. These enzymes “share the same catalytic mechanisms with their mesophilic counterparts” (Sigma, 2004). It is clear that “when cloned and expressed in mesophilic hosts, hyperthermophilic enzymes usually retain their thermal properties, indicating that these properties are genetically encoded” (Sigma, 2004). It is crucial to note that “when sequence alignments, amino acid content comparisons, crystal structure comparisons, and mutagenesis experiments indicate that hyperthermophilic enzymes are examined, they show similarities to their mesophilic homologs” (Partridge & Rees, 1995).
However, “no single mechanism is responsible for the remarkable stability of hyperthermophilic enzymes” (Partridge & Rees, 1995). It is worth noting that “increased thermostability must be found, instead, in a small number of highly specific alterations that often do not obey any obvious traffic rule” (Bell, Halling, Moore, Partridge & Rees, 1995).
It is worth noting that “after briefly discussing the diversity of hyperthermophilic organisms, the biochemical and molecular properties of hyperthermophilic enzymes are described” (Vieille et al 2001). Thermostability is one of the vital factors for enzyme kinetics. Typically, enzymes show both kinetic and thermodynamic stabilities based on the free energy of stabilization, which is characteristic of the enzyme (Kengen, Luesink, Stams & Zehnder, 1993). Thus, enzymes show different behaviors, such as melting at different and other changes in their molecular structure when temperatures are raised.
Thus, the melting temperature of an enzyme defined by an unfolding of 50% of its protein constitutes an important property of the enzyme. Typically, the energy barrier of an enzyme is the key determining factor of its kinetic stability when subjected to an environment with varying temperatures. In general, the energy barriers of the enzyme, referred to as its activation energy is usually defined on the half-life of the enzyme’s kinetic stability (Kim, Nashiru & Ko, 1996).
Thus, different enzymes are created depending on variations of their sources. Thus, each enzyme is named after the source form which it’s obtained. Examples include hyperthermophilic enzymes which are obtained from a hyperthermophile, and thermophilic obtained from thermophilic sources, among others (Benkovic & Ballesteros, 1997; Besson, Favre-Bonvin, OÕFagain & Wallach, 1995; Bucke, 1996).
It is important to note that the chemical reactions of the enzymes vary depending on a variety of factors. Research studies have shown that chemical reactions generally double with every 10 0C increase in reaction temperature. This theory is similar for both chemical catalysts and enzymes. However, since enzymes are proteins, it means they will become thermally denatured at very high temperatures, as can be seen in figure 1 (Copeland, 2000; Abu-Reesh & Faqir, 1996; Caruana, 1997; Chen, Robinson, Van Dam, Mart. Nez, Economou,& Arnold, 1991).
This experiment aims to determine the correct concentration of protein to get the assay working. Once the correct concentration is established the temperature of the water bath where the study is conducted will be increased while the storage environment is kept constant. After that, a new variable will be introduced into the experiment which constitutes increasing the storage time while the temperature is kept constant. Finally, both the storage time and the temperature will be increasing simultaneously, and observations made on the reaction rate and absorption of 440 Nm to help determine the effect of the storage time and temperature on the protease Neautrase (Drivdahl & Thimann, 1997; Guevara, Daleo, Oliva, 2001; Gupta, Beg, Lorenz, 2002; Jaenicke,& Bo¨hm, 1998).
The assay to use in the experiment is Azocasein, which is the most commonly used assay in many endo-protease enzymes that are active on casein. Such enzymes include bacterial alkaline. Azocasein has been used in previous studies in the assay of Neutrase. Azocasein is a protease substrate that is hydrolyzed in the presence of proteases. Hydrolysis of azocasein will release a dye that is red/orange in color and can then be detected by absorbance at 440nm. The reaction between the protease and the azocasein is stopped using trichloroacetic acid (TCA) (Adams & Kelly, 1998; Zacchi, Daghero, Jaeger & Eggers, 2006).
Method and Materials
Materials used in the experiment to study the effect of storage time and temperature of neutrase from bacillus amyloliquefacians included:
- Sodium phosphate buffer 100mM PH 7 1M
- Azocasein solution
- 5% TCA solution (Trichloroacetic acid)
- 0.5 M Sodium Hydroxide solution (NaOH)
- Neutrase pH 5-6.0, 1ml stock solution protease in 100mM solution
- Sodium phosphate 1M concentration, pH 6.5
- 1 M Hydrochloric acid with pH 7
- Azocasein
- Ethanol
- TCA (Trichloroacetic acid) 1M
- Sodium hydroxide in 1M
- Colorimetric spectrophotometer
Materials
A 10µl of the enzyme was added to a 100mM solution of Sodium phosphate buffer with a pH 7, which resulted in a 1ml stock solution of protease for use in the experiment. The level of concentration of the Sodium phosphate buffer was attained using a 2M hydrochloric acid. Ethanol was also combined with another solution for use in the experiment, typically by adding 2.08g of ethanol into a 50ml phosphate buffer and dissolving it in 5mg Azocasein. The environment consisted of Neutrase, a bacterially active protease, which can be sourced from respective bodies upon request. It is worth noting that the Neutrase used in the experiment has optimal chemical activities when it attains a pH value that varies between 5.5 and 7.5, on the pH scale.
Preparing the Assay
To obtain an essay with the right characteristics for the study, which works according to the chemical settings and environment, the temperature of the water bath was increased where the assay was kept while ensuring the storage environment remained the same. Later, the temperature was kept constant and other environmental variables changed. The storage environment provided incubational properties that included a temperature of 25 0C for 10 minutes.
Continue
Experimental procedure
The procedure consisted of pipetting an appropriate amount in µl of sodium phosphate buffer and Azocasein into 75ml and 125 ml solutions only. Then, the solution is centrifuged at 37oC then added to the neutrase. The centrifuged solutions are then mixed and incubated at room temperature for 10 minutes. Once the mixing is over, then, remove 100 µl aliquots from both samples and place them in suitable containers to produce 5% TCA. Then add 0.5 M Sodium Hydroxide into the resulting solution mix the centrifuges and transfer both the test and blank to suitable cuvettes. Record the absorbance at 440nm for both the test and blank in a spectrophotometer
Results
Studies based on the literature on enzymes and laboratory experiments confirm the general fact that enzymes show varying behavior in terms of thermal stability when subjected to denaturing heat and when stored over time. Typically, that is demonstrated in the excel datasheet and the resulting graphical presentation of the experiment showing the impact of heat and time on the behavior of the enzymes being investigated. Typically, the reactive nature of the enzymes is attained by placing the enzyme in a reaction medium.
Table of time against absorbance rate.
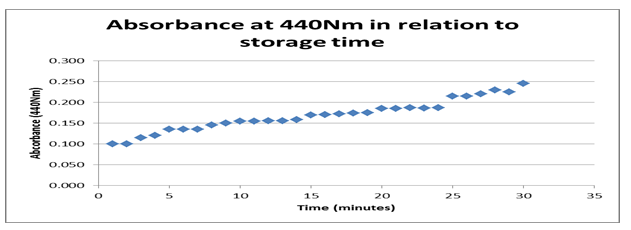
According to the findings from the datasheet and the graph in figure 1 above, the absorbance rate at440 Nm about storage time shows an optimum value of 0.25 Nm with a maximum period of 30 minutes. Typically, the graph shows a positive gradient indicating that a significant effect of time on the behavior of the enzyme is registered. However, the rise is positive between 1.0 Nm and 0.25 Nm before it terminates. That shows that time is a strong variable affecting the absorbance of the enzyme.
Typically, the datasheet shows the reaction environment that stimulated enzyme activity consisting of two variables, time and temperature. The graphs of absorbance at 440 Nm about storage time indicate a rise from 0.100 to 0.025 with a timeline between 0 and 30 minutes showing a positive gradient. The positive gradient indicates the denaturing rate for the enzyme storage, modifying its behavior with time. It confirms that enzymes in storage can be denatured even when in storage time.
Table of temperature against absorbance rate
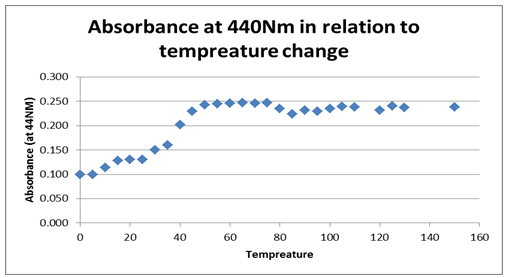
Table of time and temperature.
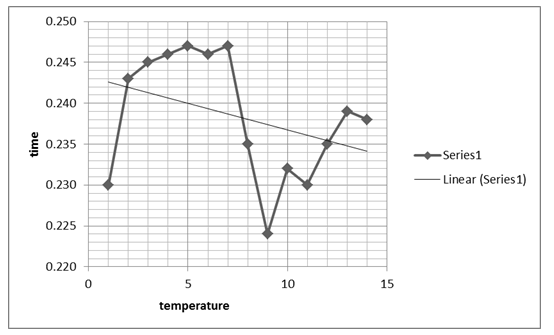
The zigzag line shows the combined effects that time and temperature have on the stability of the enzyme.
The next graph, figure 2 above, shows the relationship between temperature and the absorbance behavior of the enzyme at 440 Nm. The graph, however, shows a significant change and variation from the first graph, by indicating a positive gradient within the 0.100 and 0.025 Nm range, which changes drastically as temperature rises. At an 80 0C temperature, the graph indicates a significant drop and starts to rise again as denaturing temperature is increased, attaining a near-zero gradient with time. That implies the absorbance at 440 Nm effect becomes insignificant as temperature rises. The maximum absorbance value is attained at 0.25 Nm and at a 50 0C temperature, which seems to remain fairly constant for a range of temperature rises.
Typically, the stability of the enzyme was investigated based on the effect of temperature showing that the enzyme activity reduced significantly with temperature rise. On the other hand, enzyme activity was shown to rise irrespective of time, suggesting that time affects the activity of the enzyme.
Discussion
The laboratory results are self-evident of the denaturing effect of time and temperature on the stability of the enzymes.
The results show that different enzymes have different enzyme activity under different temperatures. Analytically, as the temperature rises, the enzyme activity rises to the optimum value before the enzyme starts to reduce its enzyme activity. It, therefore, shows that enzymes have optimal temperatures when they have high enzyme activity, which deteriorates significantly with the temperature rise. Temperature seems to be a key variable in biocatalytic processes. Biocatalytic in this case is the unique characteristic of an enzyme to retain its activeness with time.
Typically, there is an observed increase in enzyme activity when the temperature rises showing that the catalytic nature of the enzyme increases with an increase in temperature until a specific value is attained beyond which the catalytic activity remains constant. Typically, that could be due to the increase in the presence of enzyme molecules and an environment fostering the rise in chemical activities. It is possible to suggest that Proteolytic activity was determined by the reaction of azocasein with proteases in the pitcher fluid sample, which resulted in their proteolysis and the formation of colored TCA-soluble azopeptides with a high UV-absorbance. There was no enzyme activity in the controls as no enzyme was added to the reaction, therefore, the azocasein was precipitated into the pellet during centrifugation, hence why there was no absorption.
It is worth noting that the enzyme activity increases with an increase in temperature, a reaction that initially was very low but showed steep rise inactivity for the first 45 0C. Typically, that leads to the conclusion that the incubation period for the enzyme has a significant effect on its activity. It is crucial to note that as the temperature is increased further, a reverse reaction is experienced. Typically, that leads to a reduction in enzyme activity at higher temperatures. Studies show that the Neutrase start experienced a denaturing effect at higher temperatures. The denaturing process reduces the enzyme activity experienced leads to the loss of catalytic activity of the enzyme.
That is attained by the modification of the molecular structure of the Neutrase enzyme with rising in temperature. On the other hand, if the reaction temperature was increased further and measurements were taken at higher temperatures, it could be possible to arrive at false values.
The above results adduced on evidence of changes in temperature and the absorbable rate are demonstrated and explained in the thermal stability of Neutrase enzymes reaction mechanisms.
When the temperature rises in the acidic environment using hydrochloric acid, a hydrophobic effect is experienced. The hydrophobic effect causes the enzymatic molecules that constitute the proteins in the enzyme to collapse by weakening all types of bonding forces between the molecules.
These molecular bonding forces include the Van der Waals interactions and ionic bonds. A critical evaluation of the thermal process based on several theories that explain the denaturing effect of heat on the structure of the proteins shows that different types of environments have different denaturing effects on proteins. In theory, Dill (83) studied and established that nonpolar solvents denature the structure of the neutrase enzyme with a strong role played due to the hydrophobic effect on the thermal stability of the protein.
The theoretical proposition by Dill (83) was that the hydrophobic effect responsible for the structural stability of the enzyme was also responsible for the structural stability an enzyme had when under thermal investigations. Typically every scientific research and evidence point to the hydrophobic effect as being responsible for the stability of the protein when thermally evaluated. Typically, that is deductive of the stability of the Neutrase enzymes that belong to the mesophilic and hyperthermophilic homologous groups. In these groups, scientific research shows the reason for the thermal stability of the enzymes with temperature from minimum temperature to maximum temperature values.
One of the underlying reasons is that hydrophobic interactions, which are responsible for the thermal behavior of the Neutrase enzymes show a unique sequence of preserving the internal molecules better than external molecules. Another assumption in the study is that the molecules’ exposed areas have numerous stabilizing substitutions that interact with the molecules in the mesophilic and hyperthermophilic homologous groups.
Typically, the protein molecules in either of the enzyme substances are packed so effectively to leave any room for the molecules to rearrange themselves as temperature rises. The theory has it that the core of the protein during temperature changes within the allowable limit is never altered, but remains the same, thus, the chemical activity of the enzyme remains the same. On the other hand, the stabilizing effects of the enzymes are found in more exposed areas of the proteins.
However, the theory points out that a different mechanism gets involved in the stability of the enzyme with temperature variations. One of the theories points to protein amino acid composition in mesophilic and thermophilic enzymes as being responsible for the thermal stability of the enzymes. The residual content of aromatic molecules significantly influences the thermal process of stabilizing the enzymes. The higher the content of the residues, the higher the thermal stability of the enzymes and vice versa attained.
Research has shown that deamidation is another salient property relevant to the stability of the Neutrase molecules. A critical analysis of the theory behind the deamidation process indicates that the high enzymatic Neutrase activity is based on T. maritime L-isoaspartyl methyltransferase. Theoretically, it shows that the enzyme becomes highly active when the temperature rises further and further to a specified level before becoming inactive. At the height of enzyme activity, the enzyme shows high load capabilities for protein damage experienced at high temperatures. However, the variations instability, in theory, are based on key substances contained in the enzyme to enable stability at varying levels of thermal effects. These include the composition of Asn residues, among others. On the other hand, as mentioned above, aromatic interactions also evidently affect enzyme activity and behavior at varying temperatures.
Aromatic interactions constitute the interaction and distance between the phenyl ring centroids. Scientific investigations have revealed that the distance between the particles defining the possible distance of interaction between aromatic pairs is less than 7.0 A°. Scientific explanations show that aromatic pairs exist in different structures, based on molecular orientations of their core molecular structures, and their networked behavior. In theory and practice, these rings are near perpendicular to one another for the interacting pairs causing nonlocal interactions to be experienced. Studies also show that most of the structures have potential energies within different forms of residues, i.e. either buried or not buried.
During the temperature rise, aromatic interactions caused by temperature changes increase in more frequency, though the residue mentioned elsewhere in the paper plays a significant role in influencing the stability of the enzyme. Thus, the stability of the enzyme with a temperature rise is strongly associated with the structure of the molecules and the interactions between the aromatic rings.
In theory, several stabilizing features inherent in different families of Neutrase molecules have been identified. These stabilizing features are responsible for the enzyme retaining its temperature between specific values before they become denatured. These include additional intramolecular features that reinforce the stability of the enzyme, neutral H bonds with charges that enable higher molecular stabilities, the dipolar stabilization effects of the C and N caps of helices, and a significant number of prolines in loops or as may be found at the N terminus of helices.
However, the list is not exhaustive, and additional features such as minimal hydrophobic, less polar, and more charged enzyme surface as another significant feature, a significant number of aromatic residues as discussed above, and ionic interactions that occur on the surface of the molecules involved in enzyme activities. On the other hand, the structural nature of the enzymes specifically consisting of a rigid central trimer interface, and other reinforcing characteristics contribute to the thermal stability of the enzyme.
As mentioned elsewhere in the paper, Neutrase enzymes constitute different structures that contribute significantly to the behavior of the molecules. One such is the Prolines with varying levels of entropy. In theory, according to Matthews et al. (238), the stability of an enzyme can be increased by reducing its entropy and increasing the residual nature of the reactants involved in the stability and enzyme activities. A typical example, according to Matthews et al. (238) is the mutation shown as Gly→ Xaa or Xaa →Pro.
In this mutation, entropy is reduced in the process while an increase in the stability of the protein is attained. That leads to enzymes with higher thermodynamic stabilities. However, the process involves a mechanism that targets neutral protease inactivates based on a technique known as hydrolysis. In theory, it is assumed that hydrolysis constitutes a situation that targets specific surface loops to minimize the possibility of the enzyme unfolding. That leads the substance, such as the aromatic structure, to reduce the interactivity between the molecules by reducing the covalent bonds between the molecules leading to appropriate torsional angles that reinforce the stability of the substance.
It is possible to enforce certain measures to reinforce intrinsic and extrinsic stabilities on Nuetrase enzymes. In theory, enzymes falling under the hyperthermophilic group have shown very high levels of stability with temperature changes. However, other theoretical propositions point out that extrinsic factors strongly influence the stability of the enzyme. That includes environmental factors such as pressure. That is also based on the general assumption that high-temperature environments are associated with high pressures. Typically, the fact is that bacterial activities cannot occur in an environment free of temperature and pressure influences. Therefore, that reinforces the requirement for all macromolecular components of each cell in that environment to be updated to the newly attained temperature and pressure.
It is important to note, however, that pressure considerations were not made in the above experiment. However, it is also established as a matter of fact that pressure provides favorable conditions when the surface of the macromolecule its acting is small, significantly contributing to the stability of the macromolecule. Thus, at high pressures, enzymes reactively stabilized by hydrophobic interactions retain their stabilities at high pressures and temperatures. However, the converse is true for enzymes that are ionic as they become unstable at high pressures.
Storage Time
Results from the experiment show time to imply the storage of the Neutrase enzyme, a behavior that could be explained based on several underlying theories. In practice, it has been demonstrated that neutrase enzymes lose their stability with time as shown in the results above. Though the study is not exhaustive, in theory, the loss in stability is activated by changes in temperature, in this case, if the enzyme is stored in a higher temperature environment than the temperature recommended for their storage. However, that does not suggest that at the recommended temperatures, the enzyme stability remains forever.
Rather, the stability is, irrespective of the normal temperature recommended for the storage of an enzyme’s stability is affected too. Thus, the stability and time of storage depend on additional factors that are not covered here. Typically, the argument on the effects of temperature on the stability of the enzyme is based on the theory behind the denaturing effect of temperature on the chemical composition of the enzyme. Typically, time, stability, and temperature have a common toll on the stability of the enzyme.
In theory, a combination of time and thermal effects on the enzymes suggests that the three-dimensional structure of the enzyme is affected by the temperature and time variables (Haard, 1998). Thus, time is inextricably interlinked with storage temperature. Thus, time is an extrinsic factor that influences the stability of the enzymes. Thus, time and the storage stability of an enzyme theoretically draw on the strategies used in their stabilization.
In theory and practice, enzymes are known to be active in aqueous solutions that are quite favorable for the inactivation of the enzyme. Typically, the hydrous environment consisting of water molecules provides an environment favorable for the inactivation of the enzyme. According to Mozhaev, (1993), the use of additives provides an additional increase in the lifespan of the catalyst and increases the stability of the enzyme.
One of the strategies for stabilizing the enzyme over a longer storage time is the immobilization of solid carries found in the functional environment. On the other hand, an additional element that leads to thermal stability over time is based on molecular rigidity attained by covalent attachments, which has been demonstrated to be very effective. Gianfreda et al., (1985) regard the relation that causes thermal stability of the enzymes to be associated with Schiff-base linkages that occur between the proteins found in the enzymes, typically targeting the aldehyde group. In theory, the stability both with storage time and temperature calls for the manipulation of the reaction or storage medium of the enzyme.
It is, therefore, crucial to note that the molecular structures of some of the enzymes are not yet established (Wang & Jonson, 2001). However, enzyme activity is shown to increase with time, showing that Neutrase increases its activity with time. Typically, the incubation time has a strong influence on the activity of the enzymes since the activity as shown above increases with time (Vieille & Zeikus, 2001). If the reaction was allowed to continue further, thus increasing the incubation period, a decrease in the rate of product formation could be experienced, after an increase and a decrease are recorded with an increase in time.
However, the proteases start to denature and hence lose catalytic property (Knapp. Kardinahl, Hellgren, Tibbelin, Scha¨fer, & Ladenstein. 1999). It is, however, possible, deducing from the graph to obtain false values of the stability of the enzyme if the incubation period was extended. Typically, measurements taken for the enzyme activity that was measured would have been falsely low.
Conclusion
In conclusion, the experiment shows a significant effect of temperature and storage time on the absorbance of Neutrase. In practice, the enzymes become denatured as temperature rises by rearranging the molecular structure of their molecules. Typically, the molecular structure of the Neutrase enzymes influences the chemical and physical properties of the enzyme. It is the intermolecular bond and bonding characteristic that cause varying characteristics of the bonding between the molecules.
Typically, the additional intramolecular characteristics that cause the stability of the enzyme, with neutral H bonds with charges that enable higher molecular stabilities, the dipolar stabilization effects of the C and N caps of helices, and a significant number of prolines existing in loops or as may be found at the N terminus of helices in the enzyme molecules. Additional characteristics such as minimal hydrophobic concentrations, less polar characteristics, and highly charged enzyme surfaces are other significant features that influence the thermal stability of the enzymes.
A significant number of aromatic residues discussed elsewhere in the paper and ionic interactions that occur on the surface of the enzyme molecules involved in enzymatic activities have an overall implication on the thermal properties and stability of the enzyme. On the other hand, the structural nature of the enzymes specifically consisting of a rigid central trimer interface, and properties provide an additional theoretical foundation on the thermal stability of the enzyme. It is worth concluding that the temperature and time factors influence the stability of an enzyme-based on the absorbance behavior of the enzymes.
References
Abu-Reesh, I. & Faqir, N 1996, “Simulation of glucose isomerase reactor: optimum operating temperature” Bioprocess Engineering. 14:205-210.
Adams, M. & Kelly, R 1998, “Finding and using hyperthermophilic enzymes” Trends in Biotechnology, 16:329-332.
Bell, G., Halling, P., Moore, B., Partridge, J. & Rees, G 1995, Biocatalyst behavior in low-water systems, Trends in Biotechnology, 13:468-473.
Benkovic, S. & Ballesteros, 1997, Biocatalysts- the next generation, Trends in Biotechnology, 15:385-386.
Besson, C., Favre-Bonvin, G., OÕFagain, C. and Wallach, J 1995, Chemical derivation of Pseudomonas aeruginosa elastase showing increased stability. Enzyme and Microbial Technology, 17:877-881.
Bucke, C 1996, Oligosaccharide synthesis using glycosidases. Journal of Chemical Technology and Biotechnology, 67:217-220.
Caruana, C 1997, Enzymes tackle tough processing. Chemical Engineering Progress, pp 13-20.
Chen, K., Robinson, A., Van Dam, M., Mart.Nez, P., Economou, C., and Arnold. H 1991, Enzymatic engineering for nonaqueous solvents II. Additive effects of mutation on the stabilization and activity of subtilisin E in polar organic media. Biotechnology Progress, 7:125-129.
Copeland, R 2000, Enzymes, Second edition, New York, WILEY-VCH Drivdahl R.H., Thimann K.V 1997, “Proteases of senescing oat leaves I. purification and general properties,” Plant Physiol, vol. 59, pp. 1059-1063.
Fukumoto J 1943, ‘Studies on the production of bacterial amylase isolation of bacteria secreting potent amylases and their distribution’ (in Japanese). J. Agr. Chem. Soc. Japan 19: 487-503.
Guevara M. G., Daleo G. R., Oliva C. R 2001, “Purification and characterization of an aspartic protease from potato leaves”, Physiol Plant., vol. 112, pp. 321-326.
Gupta R., Beg, Q.K. & Lorenz, p. 2002. “Bacterial alkaline proteases: molecular approaches and industrial applications”, Appl Microbiol Biotechnol, 59:15–3.
Jaenicke, R., & Bo¨hm. G. 1998. The stability of proteins in extreme environments. Curr. Opin. Struct. Biol. 8:738–748.
Kengen, S. W., Luesink, E. V. Stams, A. J. & Zehnder, A. J 1993, Purification and characterization of an extremely thermostable b-glucosidase from the hyperthermophilic archaeon Pyrococcus furiosus. Eur. J. Biochem. 213:305–312.
Kim, C.-H., Nashiru, O. & Ko, J. H 1996, Purification and biochemical characterization of pullulanase type I from Thermus acidophilus. FEMS Microbiol. Lett. 138:147–152.
Kim, Y.-O., Kim, H.-K. Bae, K.-S.. Yu, J.-H & Oh, T.-K 1998, Purification and properties of a thermostable phytase from Bacillus sp. DS11. Enzyme Microb. Technol. 22:2–7.
Kimura, S., Kanaya, S. & Nakamura, H 1992, Thermostabilization of Escherichia coli ribonuclease HI by replacing left-handed helical Lys95 with Gly or Asn. J. Biol. Chem. 267:22014–22017.
Kirino, H., Aoki, M. Aoshima, M Hayashi, Y. Ohba, M. Yamagishi, A. Wakagi, T. & Oshima. T 1994, Hydrophobic interaction at the subunit interface contributes to the thermostability of 3-isopropyl malate dehydrogenase from an extreme thermophile, Thermus thermophilus. Eur. J. Biochem, 220:275–281.
Klingeberg, M., Galunsky, B. Sjøholm, C. Kasche, V. & Antranikian, G 1995, Purification and properties of a highly thermostable, sodium dodecyl sulfate-resistant and stereospecific proteinase from the extremely thermophilic archaeon Thermococcus stetteri. Appl. Environ. Microbiol. 61:3098–3104.
Knapp, S., de Vos, W. M. Rice, D. & Ladenstein, R 1997, Crystal structure of glutamate dehydrogenase from the hyperthermophilic eubacterium Thermotoga Maritima at 3.0 A°resolution. J. Mol. Biol. 267:916–932.
Knapp, S., Kardinahl, S. Hellgren, N. Tibbelin, G. Scha¨fer, G. & Ladenstein. R 1999, Refined crystal structure of a superoxide dismutase from the hyperthermophilic archaeon Sulfolobus acidocaldarius at 2.2 A° resolution. J. Mol. Biol. 285:689–702.
Sigma – Aldrich. 2011, Neutrase material safety data sheet. Web.
Skeie S 1995, ‘Influence of liposome-encapsulated Neutrase and heat-treated lactobacilli on the quality of low-fat Gouda-type cheese’ Cambridge university journals.
Vieille C., & Zeikus G 2001, Hyperthermophilic Enzymes: Sources, Uses, and Molecular Mechanisms for Thermostability. Biochemistry department. Michigan.
Wang, T., & Jonson, L. 2001, Refining High-Free Fatty Acid Wheat Germ Oil. J. Am.Oil Chem. Soc., 78, 71-76.
Zacchi, P., Daghero, J., Jaeger, P., & Eggers, R 2006, Extraction/Fractionation and deacidification of wheat germ oil using supercritical carbon dioxide. Braz. J. Chem. Eng., 23, 105-110.