Introduction
Catalyst
The use of a low-molecular weight organic molecule as a catalyst has accumulated interest in the recent years [6]. Researchers have shown that an organocatalysis reaction does not depend on metal and are both efficient and selective as metal or bio-catalysis. Meticulous work on this area has led to novel discoveries that include; the Lewis base-catalyzed enantioselective processes and a Brønsted acid organocatalysts that is likely to replace the tradition metal based asymmetric Lewis acid-catalysts [31]. The primary attraction of organocatalysis is their robust nature which includes availability, low cost, non toxic and the ability to bring novel transformations. In addition, the Organocatalyst are inert toward moisture and oxygen and demands the absence of transition metal and inert conditions. These attractive properties have led to huge research in this area. Other broadly used concepts of Organocatalyst include enamine catalysis, and asymmetric counter anion-directed catalysis [13]. The great achievements in the process development have shown that organocatalysis can be a valuable tool in the industries. Introduction Catalyst A catalyst is typically any substance added in a chemical reaction, to control either by accelerating or slowing the reaction and remains the same at the end of the reaction [44] [45]. A catalyst can either be synthetic, a metal or an organic substance. A catalyst that works to slow a process is referred to as an inhibitor and finds its importance in the pharmaceutical industry where such catalyst are used in preparation of drugs that prepare high blood pressure and mental illness. In addition, catalyst is vital in industries as they provide alternative routes that are lower in energy and hence lower activation energy. The reaction occurs when the colliding particles acquires the minimum energy (activation energy). The following diagram shows that, in presence of a catalyst more the particles undergo the chemical reaction as the energy becomes more favourable for collision.
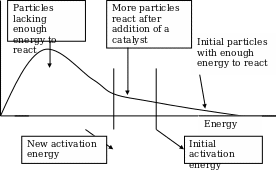
Definition of organocatalyst
Different definitions of organocatalysts is being used although they mean the same. An organocatalyst is a molecule which contains carbon, nitrogen, oxygen, sulphur, Phosphate and may also include other non metal elemements [45] [46]. The word organocatalsis is composed of two words, organo meaning organic and catalysis meaning acceleration or a process of speeding up a reaction. The word, organocatalysis, was generated in 2000 by David MacMillan from the concept of organic catalysis. There are two classes of organocatalysis, the chiral and achiral. Chiral amine catalysts and their derivatives dominate the emerging field of asymmetric organocatalysis. This technology, initially based upon the discovery of proline-catalyzed aldol reactions, has been expanded to a variety of amine catalysts, many of which have been derived from proline itself. Amine catalysts activate carbonyls by the formation of an iminium ion or an enamine.
Research is actively going on to replace organometallic with asymmetric organocatalysis. Upon discovery, organocatalysts has been used like normal catalyst until recently when the application to generate enantioselective products was advanced. Most popular organocatalysts display the secondary amine functionality. This type of organocatalysts may be illustrated to perform enamine catalysis whereby an active enamine nucleophile with excellent catalytic qualities is formed. Organocatalysts may also display iminium catalysis whereby an active iminium electrophile with catalytic quantities is formed. Typically, this mechanism describes what is referred to as covalent organocatalysis. With covalent bonding of substrate to catalyst, the catalyst usually used in the process is large quantity, for instance, proline mediated reactions requires 20-30 mol%. On the other hand, non-covalent bonding will only require low loading of the catalyst. An example of non-covalent interactions is the hydrogen bonding that requires 0.001 mol%. The metal based catalysis is being faced out slowly. Organocatalysis is widely used and making a huge contribution to green chemistry. In addition, chiral organocatalysts is open to asymmetric catalysis such as the proline mediated reactions like the aldol reactions [35]. The other group, achiral organocatalysts, is based on nitrogen atom. An example is pyridine which is widely applied in the modification of Doebner reaction of the aldol condensation. Others include DMAP and DABCO used in esterfications and the Baylis Hillman reactions, respectively. This class of achiral organocatalysis has been applied for a long time but great interest is now focusing on the chiral catalysts (asymmetric catalysis). This branch of organocatalysis is referred to as asymmetric organocatalysis or enantioselective organocatalysis.
In recent years, the demand of chiral molecules has increased considerably and intensive research has been elicited in order to develop new advanced methods to push the limits of synthesizing such compounds. In the past, enantiomerically enriched compounds were generated either via classical resolution or by chemical transformation of an enantiomer precursor obtained with the help of chiral auxiliaries. The enantioselective synthesis using auxiliaries requires stoichiometric amounts of chiral substances which are not incorporated in the target molecule and have to be removed after the sterogenic centres are introduced. Meticulous work has been noted in asymmetric transformations where the achiral starting materials are converted into enantiomerically enriched products with the help of a chiral catalyst. However, effective catalysts used in several processes are readily available in nature such as the biocatalysts while others are designed and synthesized in the laboratories and are referred to as chemical catalysis. Another category of catalyst is enzymes. They occur naturally and are highly selective while conducting the reactions. However, they have a limitation in that only one enantiomer of the product can be obtained directly. In recent years, much effort has been made in the field of organomettalic catalysis. A major progress was noted in the early 70’s by Knowles and his colleagues who were the first to perform enantioselective hydrogenation of prochiral olefin that constituted of 96% ee in the presence of rhodium-based catalyst. The advance process was eventually commercialized and applied on a large scale in the synthesis of anti-Parkinson drug L-DOPA (95% ee). Later, Knowles shared a Nobel Prize (2001) in chemistry with Noyori for enantioselective hydrogenation and Sharpless for enantioselective oxidation catalysts [46] [49]. Most importantly, all these processes have found great use in industries in the manufacture of chiral drugs and building blocks for asymmetric synthesis [46]. The concept of asymmetric catalysis is connected with that of metal-complex Catalysed syntheses and much work has focused on the design and synthesis of organometallic complexes which are able of inducing asymmetry during the transformations. Recent research is now focusing on the use of simple organic molecules to induce chirality in the products. This is referred to as organocatalysis. In reality, most popular organocatalysts are widely known ligands in the field of organometallic chemistry. This has contributed to a major breakthrough on the modern organic synthesis and the field of organocatalysis is on rapid growth. The widely used catalysts include the amino acid and derivatives, small peptide chains, cinchona alkaloids as well as chiral diamines and heterazolium based compounds. The range of organocatalysis has actually been extended considerably and a large group of reactions are carried out efficiently in the presence of these small chiral molecules [16]. Their potentiality was confirmed in the last three years when a typical-metal mediated coupling reactions was performed under metal free conditions. An Organocatalyst is typically a pure organic, metal free substance that is used to speed up the rate of a reaction. The Organocatalyst constitutes of small molecules that are easily designed and synthesized from nontoxic compounds such as sugars, peptides or even amino acids [17].
Major classes of organocatalysts
In most practical cases, organocatalysis is applied through asymmetric synthesis. This is further categorized in various classes. The class of biomolecules includes; proline, the cinchona alkaloids, phenylalanine and may also have certain oligopeptides. A number of catalysts have been developed synthetically from this biomolecules. This includes McMillan imidazolidinones and the CBS catalysts derived from proline. The other class is composed of the derivatives of binol such as the nobin. The triazolium salts catalyzes the next generation of Stetter reaction. Finally, the class of organocatalysts based on thioureas is also popular in most leading industries [46]. There are a number of asymmetric reactions involving the application of organocatalysis. The asymmetric Diels-Alder reaction also commonly known as cycloaddition is a reaction involving a substituted alkene and a conjugated diene (specifically, a dienophile) to generate a substituted cyclohexene system. This type of reaction is an example of pericyclic reactions which occurs through a transition state to form product [10] [46]. Asymmetric Michael additions consist of a group of reactions that have been focused on in recent times. Commonly applied methods include the chiral phase transfer catalysis that involves the chiral quaternary ammnonium salts [7] [14]. The ammonium salts are derived from the cinchona alkaloids and proline in organocatalysis. Another widely used asymmetric organocatalysis is the Mannich reactions. The asymmetric Mannich reaction is a widely applied means of producing beta amino carbonyl compounds starting from cheap and readily available substrates [21] [22]. In this reaction an aldehyde, an amine, and a ketone react in a three-component– one-pot synthesis. In this reactions two prochiral centers usually results that gives rise to two diastero-meric pairs of enantiomers. As a synthetic alternative, the reaction can also be performed as a nucleophilic addition of a carbon nucleophile to a preformed imine which is prepared starting from the aldehyde and an amine group [15]. An asymmetric Mannich reaction was recently successfully achieved by means of a wide variety of catalyst including the metal and the organocatalysts. With the latter catalyst the reaction may be performed asymmetrically by use of naturally occurring L-proline and other related compounds such as chiral organocatalyst. A key advantage of the proline-catalyzed route is that unmodified ketones are used as donors, which is synthetically highly attractive. In contrast, many other asymmetric catalytic methods require preformed enolate equivalents as nucleophile. The Shi epoxidation is also an example of asymmetric organocatalysis. The reaction involves a reaction between olefins with oxone (potassium perxymonosulfate) with a catalyst derived from fructose. The reaction produces epoxides with a high enantiomeric excesses characteristic from trans disubstituted alkenes and trisubstituted alkenes [52]. Examples of Shi epoxidation products include the cis-disubstituted alkenes and the styrenes.
Thiourea organocatalysis Generally, the hydrogen bonding or the non-covalent plays a vital role in catalysis that is highly characterized by selective molecular recognition, substrate activation and numerous organic transformations [23]. Both theoretical (computational) and experimental have been carried out by a number of investigators of hydrogen bonding ability in thiourea compounds. It was established that simple reactions of Diels-Alder often act as Lewis acid [60]. They act explicitly through a mediated hydrogen bonding rather than the covalent bonding. The most effective achiral thiourea catalysts is the N,N’-bis[[3,5-bis(trifluormethyl)phenyl thiourea and consists of excellent organocatalysis features such as being electron deficient, has a rigid structure and the preferred substituent is trifluoromethyl group. In general thiourea derivatives have numerous advantages [29]. This include, they are non product inhibition, low catalyst loading, inexpensive and simple procedures in preparation, green chemistry, inert in nature, water friendly and non toxic. Thiourea derivates organocatalysts have been widely used to date due to the above advantages. This also forms a class of hydrogen bonding organocatalysis.
Finally hydrogen bonding plays a vital role in organocatalysis techniques. The lone pairs of the carbonyl oxygen atom are considered as hard Lewis base sites. When the coordination of these lone pairs to Lewis acids occurs, the electron density at the oxygen atom and the lowest unoccupied molecular orbital (LUMO) that activates the group toward nucleophilic attack is lowered. Hard, small Lewis acids are especially effective in carbonyl activation. The proton, being the smallest possible Lewis acid, is one of the best. However a plethora reactions involving carbonyl group can react in protic acid medium. Surprisingly, these reactions suffer badly [51]. This includes destruction of requisite nucleophiles such as the enolates, enamines and cyanides. Furthermore, protic acid induces side reactions such as elimination, epimerization and polymerization procedures. For these reasons, carbonyl activation by hard Lewis acids has become the mainstream approach towards selective carbon–carbon bond formation in, for example, aldol condensations, and asymmetric Diels– Alder and hetero-Diels–Alder reactions [1].
Strong protic acids are not required as catalysts if the reaction proceeds under general acid catalysis, that is, the proton transfer occurs only in the transition state or the catalyst stabilizes the transition state by hydrogen bonding. Recent research has elicited that the role of carboxyl group of L-proline is to activate the carbonyl acceptor by hydrogen bonding. Moreover on the same note, it has been demonstrated that an excellent aldol catalysts rely on the double hydrogen bonds in activating the carbonyl acceptors.
Benefits of organocatalysts
Organocatalysis offers a harmonious environment in well established chemical and biochemical transformation and most of their practical advantages are notable. The organocatalysts are inexpensive and are more stable than the metal based complexes. In addition, the reactions generally tolerate aerobic conditions and do not require absolute solvents. Organocatalysts have demonstrated a wide scope of substrate such as enantiomeric form of the chiral catalyst and are also easily synthesized. In addition, the organocatalyst can easily modify the substrate by simple chemical reactions than their counterparts. Another advantage worth of noting is that, these catalysts are easily linked to a solid support which contributes to their wide usage in the industries. In addition, they are easily recovered after the transformation and repeatedly recycled without any loss of selectivity. On the other hand, organocatalysts have one limitation in that a large amount of the catalyst is often used in a single reaction [6] [32]. In this concept of the organocatalysis, the transition state necessary for enantioselective transformation is achieved through passive or dynamic interactions unlike in the case of metal-mediated enantioselective catalytic reactions where the metal is the one responsible for transformation. In the dynamic bindings, the substrate is activated through nucleophilic addition of the chiral amine catalyst to the prochiral substrate. Such bindings also depend on the formation of hydrogen bonding between substrate and catalysts to help in the stereo-selectivity processes. A passive interaction includes hydrophobic, van der Waals and electrostatic interactions. Both dynamic and passive interactions, display characteristics features and mechanistic similarities to known bioorganic catalysts are referred to as enzyme mimetics. So far, this class of catalysts has displayed effective results in a wide range of substrates in various reactions and therefore the need arises to treat them as highly privileged class of ligands.
Examples of Organocatalyst include: proline, cinchona alkaloids, and other secondary amine derivatives which are shown below.
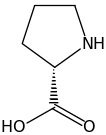
Proline and its derivatives effectively catalyze different types of reactions such as intra- and intermolecular aldol reactions, Michael addition, Mannich reaction, [4+2] cycloaddition, Baylis-Hillman reaction [7] [27], as well as alkylation and amination of aldehydes. Cinchona alkaloids have enlarged the field of organocatalysis and extraordinary results have been achieved in reactions involving anhydride desymmetrization, conjugate addition, ketene chemistry and kinetic resolution.
Comparison with conventional catalysis
The scope of organocatalysis has been extended considerably and a large range of reactions are now performed efficiently in the presence of these small chiral molecules. Organocatalysis now offers complementary approaches to the well-established chemical and biochemical transformations and some of their practical advantages are notable [59]. The catalysts are inexpensive, easily available and usually more stable than the metal-based complexes. Organometallic catalysis has a wide substrate scope and high catalytic activity. However, this class of catalysis is limited by the fact that the process involved is tedious and pollution of the environment by heavy metal. Similarly, enzymatic catalysis has both advantages and disadvantages. This class has demonstrated a high selectivity and catalytic activity though limited by low substrate scope. The understanding of mechanistic pathways of organocatalysis is improving since this area has accumulated more interest. This reaction is suggested to proceed via a loose transition state mediated by chiral metal complexes. This class is associated with a high level of enantioselectivity through hydrogen bonding or ion pairing. The transition-metal-mediated coupling reactions such as Tsuzuki and Heck-type as well as Tsuji-Trost reaction can now be carried out under metal-free conditions [56]. Metal centers act as Lewis acids and organic catalysts tend to react as heteroatom-centered mainly composed of N(O)-, P(O)- Lewis bases. Novel privileged group of unexplored catalysts such as asymmetric catalysis by Bronsted acids is emerging. A wide variety of developments focuses the use of synergic systems and bi-function catalysts with two functionalities such as Lewis base and Lewis acid. Organocatalysis have eventually grown and moving toward maturity. Nowadays, it has found use outside the academic environment to pharmaceuticals industries such as Pfizer. Merck, AstraZeneka, GlaxoSmithKline among others. The great practical potentiality is linked to their advantage mentioned above.
Literature review
Classification of Organocatalyst
The classification of Organocatalyst is based on the mechanism of catalysis namely Lewis bases, Lewis acids, Brønsted bases and Brønsted acids [60]. The four categories of catalysis are explained below using schematic diagram;
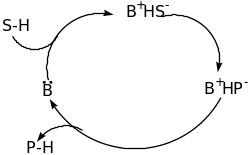
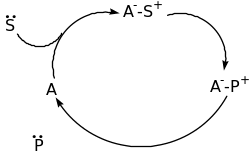
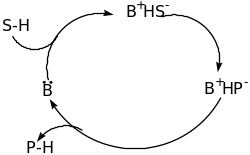
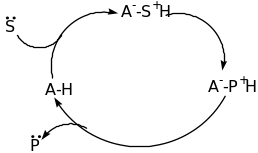
From the above schematic diagram, it is clear that the Lewis base catalyst (B:) initiate the catalytic cycle via nucleophilic addition to the substrate (S). this results into a complex that releases the product (P) and the catalyst is reused. For the Lewis acid catalysts (A), activation via nucleophilic substrates (S:) takes place. Brønsted base and acid catalytic cycles are initiated via a (partial) deprotonation or protonation, respectively. The four types of organocatalysis have been elaborated further in the following paragraphs.
Lewis base catalysis
The concept of the Lewis base catalysis can be formulated from the following broad definition. The process involves an electron pair donor that accelerates the rate of a chemical reaction by interacting with an acceptor molecule in one of the substrates. Typically, the Lewis base catalysis is effective in promoting the reactions of nucleophilic, electrophilic and ambiphilic reagents. Regardless of the nature of the Lewis base, the acceptor fragment within the adduct possesses increased electron density relative to the parent Lewis acid. The distributions the electron density among the participating atoms must be considered to rationalize the effects of adduct formation on reactivity.
Enamine catalysis
In this type of catalysis, a catalytically generated enamine intermediate reacts with a wide variety of electrophiles or undergoes pericyclic reactions. The first example was demonstrated by Hajos–Parrish–Eder– Sauer–Wiechert reaction. The mechanisms involving this reaction was poorly understood and revival of this chemistry was initiated by the discovery of Proline catalyzed direct asymmetric intermolecular aldol reaction in recent years [18] [24]. This was the first reaction that pioneered this area of organocatalysis and was developed by two pharmaceuticals companies namely Hoffman-La Roche and Schering Plough [27] [28]. The aldol reaction involved the chiral proline catalyst which occurs naturally. The reaction is initiated 3 mol% that accelerates achiral triketone to generate a product of 93% enantiomeric excess of ketol [13]. Since more research has been focusing on proline catalyzed enantioselective intermolecular aldol reactions and the Michael additions [7] [42]. In addition, this development has also been extended to highly enantioselective α-functions of aldehydes and ketones such as aminations, alkaylitions and chlorination, bromination and fluorination using L-proline and chiral imidazolidinones as catalyst [11] [25].
Brønsted Acid – Base
In general, a Brønsted acid is defined as a substance which can donate a proton whereas a Brønsted base accepts a proton or hydrogen ion [57]. A typical example illustrates the Brønsted Acid – Base interaction;
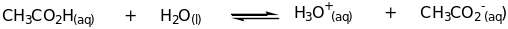
Brønsted Acid Organocatalyst
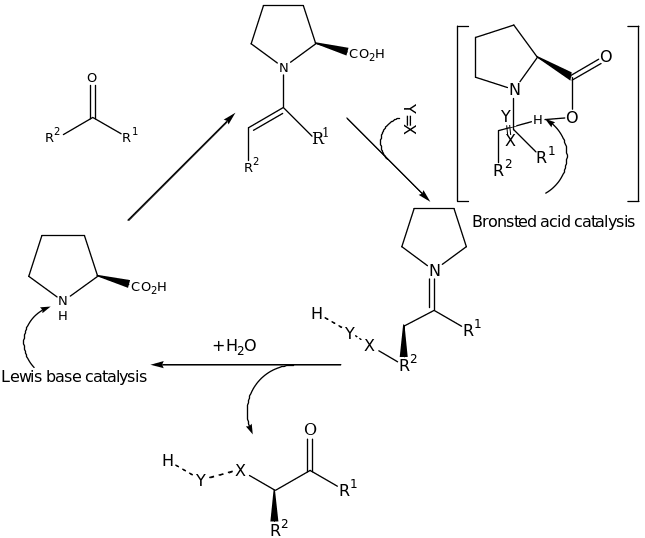
This type of organocatalysts covers interesting recent findings. Proline plays a dual role in the proline-based enamine catalysis. The amino group of proline acts as Lewis base whereas the carboxylic group acts as a Brønsted Acid as illustrated in scheme 1 [19] [57].
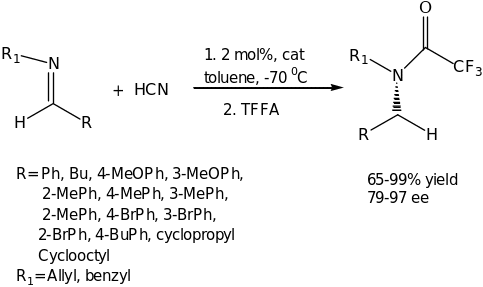
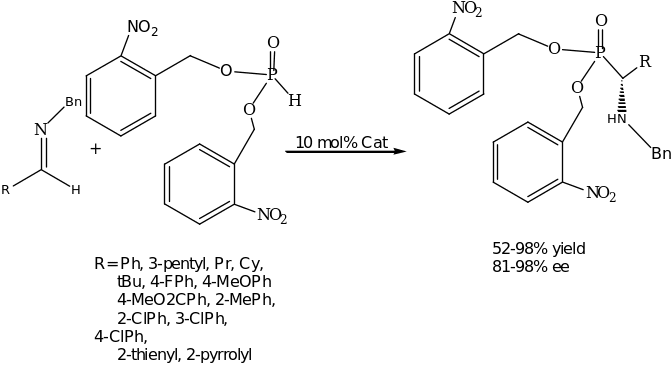
The most striking reaction under the umbrella of Brønsted Acid Organocatalyst is the asymmetric catalytic Strecker reaction which demonstrates an elegant approach for the synthesis of optically active α-amino acids. In this reaction, an active organocatalysts was developed by Jacobsen group namely optically active urea or thiourea derivatives. This discovery presented remarkable catalysts that were efficient to date in carrying out asymmetric hydrocyanation of imines. The reaction is clearly illustrated in scheme 2. In the presence of the readily available organocatalyst with a catalytic of 2 mol% the asymmetric hydrocynation proceeds with high conversion and enantioselectivity for a wide range of imines. The product, α-amino nitrile is conveniently isolated as triflouroacetamides which can further be converted into a variety of enantiomerically pure building blocks such as the amino acids.
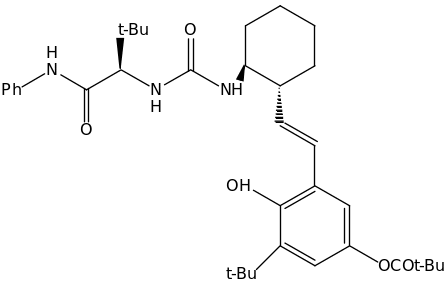
Catalyst
From the above reaction, the aromatic aldimines (R= Ar and R” = H) generated products with up to 99% ee whereas the aliphatic groups and electron deficient (R= 3-nitrophenyl) as well as heteroaromatic derivatives afforded a racemic mixture (products). This problem was later solved by synthesizing new peptide-based catalysts and optimizing them through combinatorial chemistry. The two ligand libraries consisted of 48 + 132 members that were screened in the asymmetric Strecker reaction. The excellent results were achieved with Urea and thiourea derivatives (Scheme 3). The remarkable catalyst has been applied successfully in a broad range of aryl and alkyl aldmine substrates as well as various ketimines.
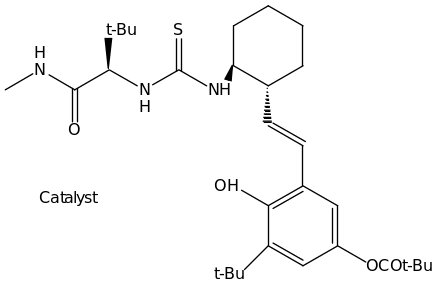
The great achievement of the organocatalytic Strecker Reaction was demonstrated by the synthesis of two optically active amino acids (Scheme 4). In this reaction unnatural (R)-tert-leucine and (R)-(-methyl phenylglycine were isolated in good overall yields with >99% ee.
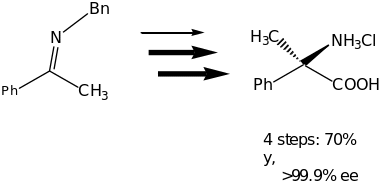
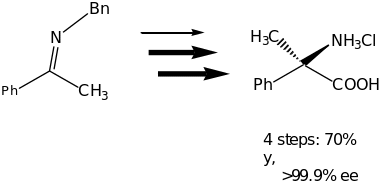
Brønsted Base Organocatalyst
In the recent years, a lot of energy has been used in development of general and selective desymmetrization methods [30]. The novel concepts for the ring opening reaction involving carbonbased nucleophiles have so far been reported. Detailed mechanistic pathway has described by Rovis on the alkylation of cyclic meso-anhydrides catalyzed by a nickel complex. In addition, he tested several phosphine ligands in combination with Ni(COD)2 as well as diethylzinc to cyclohexanecarboxylic anhydride. The mechanism involves several steps. Firstly, an oxidative addition steps between the low-valence nickel complex and the electron deficient C-O bond of the anhydride occurs. The resulting organometallic complex undergoes a transmetalation with the zinc reagent to provide the key species [20]. This follows a reductive elimination to generate the product and regenerate the catalysts. Moreover, the yield increased considerably when electron-deficient olefins were used as promoters for reductive elimination. Scheme 5 outlines the catalyst for this reaction and an example of desymmetrization reaction.
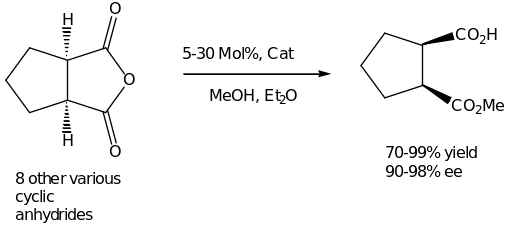
Lewis acid catalysis
Generally, the Lewis acid catalysis has been a great interest in organic synthesis. The reactions involving this catalysis must be carried out under strictly anhydrous conditions. The presence of water stops the reaction because most Lewis acids immediately react with water rather than the substrates and end up decomposing. This, however, have restricted the use of Lewis acids in organic synthesis. On the other hand, it is desirable to use water as the reaction solvent since water is safe, harmless and environmentally friendly solvent. This has led to development of water stable Lewis acids. This includes; novel metal salts (such as rare earth metal triflates) and a Lewis acid-surfactant-combined catalyst. The activation in Lewis acid organocatalysis is achieved by either an iminium ion or ion pairs. The condensation of a carbonyl compound with a secondary amine leads to the formation of an iminium ion whose LUMO has low energy after conjugation with alkene or an aromatic ring. This particular reaction has a number of applications such as in the transformation of cycloaddition or alkylation reactions in the presence of electron rich aromatic rings or nitro compounds. However, research has shown the likelihood of these traditional metal-based organocatalysts being faced out by the above described Brønsted acid organocatalysts in years to come [57].
Proline
Research has proved that, until now the most successful catalyst for enamine type reactions is L-proline. The natural L form is commonly used although both enantiomers of proline are widely available. This however marks an advantage over enzymatic catalysis. The simple amino acid can carry out reactions of the same level as the complex machinery of the natural enzymes. Proline is a natural amino acid with secondary amine functionality and has a higher pKa value and enhanced nucleophilicity relative to other amino acids. Proline can therefore react as a nucleophile with carbonyl groups or Michael acceptors to form iminium ions or enamines. The carboxyl group of the amino acids acts as Bronsted acid in these reactions; proline is regarded as a bifunctional catalyst [5]. Proline molecule forms a highly organized transition state with extensive hydrogen bonding networks promoting its exception in high enantioselectivity. In all proline-mediated reactions, the hydrogen transfer from the amine or carboxylic group of the proline to formation of alkoxide or imide is fundamentally important for charge stabilization and C-C bond formation in the transition state. However, proline-mediated reactions are been challenged in the dimerization or oligomerization of unbranched aldehydes [15]. Reactions with acetaldehydes have demonstrated vey low yields and low selectivity. Recent research has shown that, chiral imidazolidinone offers better rates and selectivity compared to proline when reacted with high complex oligopeptides. The first successful attempt to use organic molecules as chiral catalysts was noted in the early 1970s by Hajos and Parrish and Eder, Sauer and wiechert who worked independently [24]. The two groups established that, asymmetry could be induced through Robinson type annulation of an achiral triketone by simply adding a considerable catalytic amount of D- or L- praline to the mixture[2]. This is demonstrated in scheme 1 below.
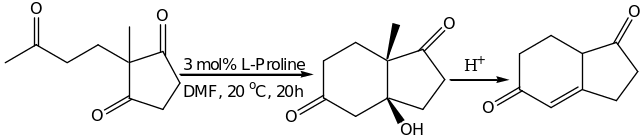
This reaction was discovered in 1970 and it formed the basis of organocatalysis. The Hajos and Parrish group catalytic process led to optically active ketol. The resulting product was later modified by Eder, Sauer and Wichert which gave an optically active dione. This was landmark discovery and it paved way to asymmetric organocatalysis.
In conclusion, we can suggest that proline is a versatile organic catalyst capable of promoting a variety of useful transformations including many which are enantioselective in nature. Both enantiomers of proline are available and this allows some flexibility. The catalyst is inexpensive, commercially available and a valuable green chemistry commodity. Many of the procedures involving proline can run at room temperature with even wet solvents. The processes are practically very simple and the conditions are usually specific per transformation.
Aldol condensation
The intermolecular version of a cross-aldol reaction was first developed by List, Lerner and Barbas [8] [36] [37] [39]. The reaction involved a reaction between aldehydes as acceptors and ketones as donors using catalytic amounts of proline [48]. The aromatic aldehydes gave products with moderate selectivity of approximately 70% ee. In addition to their findings, a variety of mono and di-substituted aliphatic aldeydes provided aldols with outstanding enantioselectivities of up to 99% ee as shown in scheme 2 below [33].
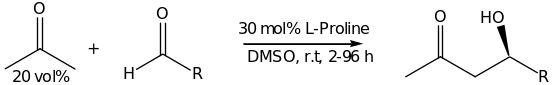
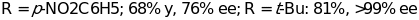
Further research gave 99% ee when unbranched aliphatic aldehydes were used in optimized conditions. This is however advancement in the cross-aldolization process which was earlier hindered by a process of self condensation and later improved by carrying out the reaction in optimal conditions (i.e. the temperature of the reaction should be kept at 4 0C) [48]. List’s work is consistent with Houk’s theoretical calculations and the proposed metal-free Zimmerman_traxler-type transition state which is catalyzed by one molecule of proline in the same way as type I-aldolases and the catalytic antibodies or their mimics [37] [38] [42]. Accumulative research and findings by MacMillan and Kunz, in addition, shed more light in proline-catalyzed reactions [59]. More substrates demonstrated that unbranched aldehydes can be used as donors in the reaction with aldehyde acceptors giving products in high yields of up to 99% ee. On the same note, Cordova reported the direct asymmetric cross-aldol reaction of aldehydes in an ionic liquid medium leading to optically pure 3-hydroxy aldehydes of about 99% ee and good diastereoselectivities [18] [48]. Fascinating results on the asymmetric intramolecular version of Hajos-Parrish-Eder-Sauer-Wiechert reaction has been noted together with the enolexo aldolization of pentane-1, 5- dialdehydes which resulted in high yields products and diastereoselectivities, with up to 99% ee. More research on L-proline confirmed that other types of reaction in a highly selective manner are possible [9]. For instance, the proline catalyzed Mannich reaction offers a useful reaction in enantiomerically enrinched amino carbonyl compounds [22]. In optimal conditions, it can be carried out directly, as a three-component one pot reaction or indirectly using preformed imines and enolates. Various ketones and aldehydes have demonstrated excellent selectivities and products of high yields when reacted with different aniline derivatives. These reactions follow an enamine mechanistic pathway and occur with diastereo-and enantioselectivity contrary to that of related aldol reaction [33]. However, when L-proline was used as a catalyst in the asymmetric Michael addition of ketones to olefins and alkylation of aldeydes, only modest levels of enantioselection were achieved. On the other hand, tremendous yields were also realized when L-)-methyl proline (10 mol%) was employed in the intramolecular alkylation reaction [42]. Also note of worthy is the independent work of MacMillan, Hayashi, Zhong who recently reported on the proline catalyzed enantioselective)-oxyamination of aldehydes illustrated in scheme 3;
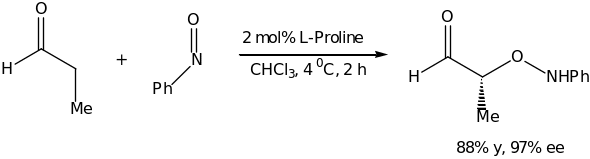
Accordingly, Cordova reported excellent chemo-, region- and enantioselectivities in the oxymination of cyclic as well as acyclic ketones. This key reaction is illustrated in scheme 4.
Amino acid derivatives
Imidazolidinone derivatives
Chiral amines such as cinchona alkaloids, ephedrine and prolinol derivatives are organocatalysts that are known to catalyze reactions with a moderate level of enantioselectivity. MacMillan reported the first highly selective Diels-Alder reaction between unsaturated aldehydes and various dienes which employed the use of secondary amines as the chiral catalysts [4]. The concept was based on the likelihood of lowering the LUMO energy of the dienophile upon a reversible formation of an iminium ion. This process is analogous to the Lewis acid catalyzed Diels-Alder reaction the activation of unsaturated carbonyl occurs upon chelation and is referred to as iminium catalysis [34] [51]. The carbonyl compounds forms iminium ions with primary and secondary amines leading to high susceptibility to react with nucleophilic species. The low LUMO energy are as a result of empty π* orbital of the conjugated double bond. This further contributes to high reactivity of the iminium ions of both aldehydes and ketones. The general mechanism of iminium activation is outlined in scheme 5 below.
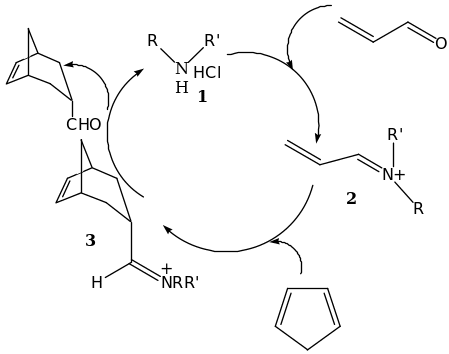
The iminium ion 2 is formed from a reaction between a catalyst and aldehyde. This is then followed by the Diels-Alder cycloaddition to form the iminium ion 3. A subsequent hydrolysis provides the enantiomerically enriched product and liberates the catalyst [4].
In conclusion, the organocatalysis of asymmetric Diels–Alder reaction is a highly efficient process, in particular when using TADDOL as well as the imidazolidinone type of catalysts that were developed by the MacMillan group. In this connection a specific highlight is certainly the application of this concept in the first highly enantioselective catalytic Diels–Alder reaction with a, b-unsaturated ketones as dienophiles. Furthermore, suitable organocatalyst have been found for the hetero-Diels–Alder reaction as demonstrated by the Jorgensen group.
Cinchona alkaloid
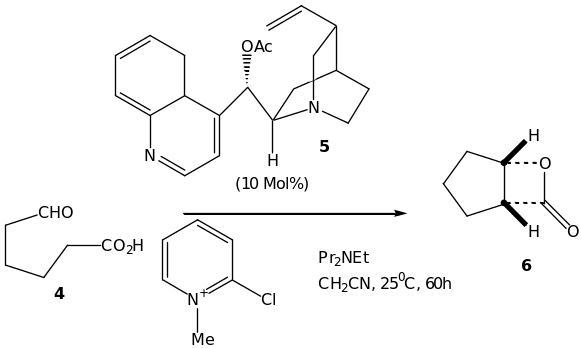
Our next privileged class of catalysts and ligands for asymmetric synthesis are the natural occurring cinchona alkaloids. They are the most widely used in the pharmaceutical, chemical and food industry. Their family consists of four pairs of pseudoenantiomers: quinine (QN) – quinidine (QD), cinchonidine (CD) cinchonine (CN) as well as the corresponding hydrogenated analogs. These classes of alkaloids are readily availability, stable and are inexpensive. They are also efficient phase-transfer catalysts for asymmetric catalysis. Their widely usage is associated with their advantage in that they posses a naturally occurring (pseudo) enantiomeric counterpart and that their structure can be easily modified in order to obtain a more effective catalyst. More surprisingly, limited information is known on either electronic nor the steric factors that determine the enantioselectivity. An example of a cinchona alkaloids catalyzed reaction is outlined in scheme 6. It is an asymmetric intramolecular aldol reaction of 4 catalyzed by the O-acylated cinchona alkaloid 5 to form an aldol product. The product formed then undergoes cyclization in situ to form final product 6, the bicyclic b-lactone. In addition, a range number of esters and carbonates of the parent cinchona alkaloid is suggested to lead to similar enantioselectivities [41].
Outlook
Numerous reactions of asymmetric organic reactions can be accelerated by a considerable amount of a chiral organic molecule [43] [44] [45]. This novel discovery in organic chemistry area has emerged as a major breakthrough and is currently celebrating its golden period. Despite the importance of substrate dependence earlier discussed, more and more transformations now meet the standards of established asymmetric reactions. Scientists now understand the basic factors that control reactivity and selectivity in these reactions. However, the rational design of catalysts in most cases has not been attained. The number of Organocatalyst is on the rise which has increased the development of enantioselective reactions tremendously. As new asymmetric reactions emerge, the success has been due to automization and computational techniques that have facilitated both the discovery of catalyst and screening of reactions for catalysts of the next generation.
Conclusion
The use of metal-free, low molecular proline-based catalyst was first reported in 1971. The reaction involved an intramolecular asymmetric aldol reaction of triketone by two groups of researchers. For three decades the area of organocatalysis was ignored and it blossomed recently. Selected recent developments in the area of asymmetric organocatalysis have been briefly described in this work. The number of researching groups in organocatalysis is rapidly increasing. Besides the fact that numerous work and development have been achieved, organocatalysis topics have been regarded to be at their infancy stage. However, the time of infancy is over and its clear from the topic described above to put organocatalysis in the bracket of adolescence. In recent years, the scope of organocatalysis covers from the generation of complex molecular systems to considerable technical synthesis process. Mechanistic pathways elaborating the fundamental procedures have been established and this generates confidence in the success of many challenging accomplishments that rely on the organocatalysis key steps [12] [13]. The impression created by this class of catalysts is visible. All modern organic chemistry textbooks have clearly illustrated the discovery of asymmetric synthetic methods that are promoted by metal complexes with chiral organic ligands [35]. The excellent progress is pointed by the wide scope of transition-metal catalyzed coupling reactions, asymmetric hydrogenation of olefins and osmium-catalyzed asymmetric dihydroxylation of olefins. These methods formed the landmark as they made many once impossible syntheses possible [47]. Additionally, more advantages are been experienced compared to organometallic and enzymatic catalysis [12]. The high cost involved in preparation, at the likelihood of contaminating the final product as well as the lack of orthogonality with a broad range of organic molecules can not further be ignored. These problems are being solved by using organocatalysts rather than organometallic. Further, figure 2 below highlights the maturity and vastness of organocatalysis.

At this age bracket of maturity, the need arises for the advancement of organocatalysis particularly the creation of new structure classes with a wide range of chemical and stereo-chemical properties. Organocatalysis is basically the key step of organic synthesis as it allows the basic structure of the molecular fragments to be modified efficiently. The sermon of this class of catalysis is evident in its application in total synthesis of natural products and biologically active molecules.
References
Agami, C. and Puchot, C. (1986). “Kinetic analysis of dual catalysis by proline in an asymmetric intramolecular aldol reaction.” Journal of Molecular Catalysis, 38, 341–343
Agami, C., Meynier, F., Puchot C. et al. (1984). “New insights into the mechanism of the proline-catalyzed asymmetric Robinson cyclization; structure of two intermediates. Asymmetric dehydration.” Tetrahedron, 40, 1031–1038
Agami, C., Meynier, F., Puchot, C. et al. (1984). “New Insights into the Mechanism of the Proline-Catalyzed Asymmetric Robinson Cyclization; Structure of Two Intermediates. Asymmetric Dehydration.” Tetrahedron, 40, 1031-1038
Ahrendt, K., Borths, C. and MacMillan, D. (2000). “New strategies for organic catalysis: The first highly enantioselective organocatalytic Diels–Alder reaction.” Journal of American Chemical Society, 122, 4243–4244
Akiyama, T., Itoh J. and Fuchibe, K. (2005). “Chiral Brønsted acid catalyzed enantioselective hydrophosphonylation of imines: asymmetric synthesis of alpha-amino phosphonates.” Organic Letters, 7, 2583–2585
Albrecht, B. and Gröger, H. (2005) Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis. New York: Wiley.
Alexakis, A. and Andrey, O. (2002). “Diamine-catalyzed asymmetric Michael additions of aldehydes and ketones to nitrostyrene.” Organic Letters, 4, 3611– 3614
Allemann, C., Gordillo, R., Clemente F. et al. (2004). “Theory of asymmetric organocatalysis of Aldol and related reactions: rationalizations and predictions.” Accounts of Chemical Research, 37, 558–569
Bahmanyar, S. and Houk, K. (2001a). “The origin of stereoselectivity in proline catalyzed intramolecular aldol reactions.” Journal of American Chemical Society, 123, 12911–12912
Bahmanyar, S. and Houk, K. (2001b). “Transition states of amine-catalyzed aldol reactions involving enamine intermediates: theoretical studies of mechanism, reactivity, and stereoselectivity.” Journal of American Chemical Society, 123, 11273–11283
Beeson, T. and MacMillan, D. (2005). “Enantioselective organocatalytic alpha fluorination of aldehydes.” Journal of American Chemical Society, 127, 8826– 8828
Behr, A. (2002) Organometallic Compounds and Homogeneous Catalysis. Weinheim: Wiley-VCH.
Berkessel, A. and Gröger, H. (2005) Asymmetric organocatalysis. Weinheim: Wiley-VCH.
Betancort, J. and Barbas, C. (2001). “Catalytic direct asymmetric Michael reactions: Taming naked aldehyde donors.” Organic Letters, 3, 3737–3740
Brown, S., Brochu, M. and MacMillan, D. (2003). “The direct and enantioselective organocatalytic alpha-oxidation of aldehydes.” Journal of American Chemical Society, 125, 10808–10809
Caddick, S. and Jenkins K. (1996). “Dynamic resolutions in asymmetric synthesis.” Chemical Society Reviews, 25, 447–456
Cheong, P. and Houk, K. (2004). “Origins of selectivities in proline-catalyzed alpha aminoxylations.” Journal of American Chemical Society, 126, 13912– 13913
Córdova, A., Watanabe, S., Tanaka, F. et al. (2002). “A highly enantioselective route to either enantiomer of both alpha- and beta-amino acid derivatives.” Journal American Chemical Society, 124, 1866–1867
Denmark, S., Gregory, L. and Beutner, A. (2008). “Lewis Base Catalysis in Organic Synthesis.” Angewandte Chemie International Edition, 47, 1560 – 1638
Elschenbroich, C. (2006) Organometallics. Weinheim: Wiley-VCH
Enders, D. and Seki, A. (2002). “Proline-catalyzed enantioselective Michael additions of ketones to nitrostyrene.” Synletters, 2002, 26–28
Enders, D. and Vrettou, M. (2006) “Asymmetric synthesis of (+)-polyoxamic acid via an efficient organocatalytic Mannich reaction as the key step.” Synthesis, 13, 2155–2158
Grieco, P. A. (eds.). (1998) Organic Synthesis in Water. London: Blacky Academic and Professional.
Hajos, Z. G. and Parrish, D. R. (1974). “Asymmetric synthesis of bicyclic intermediates of natural product chemistry.” Journal of Organic Chemistry, 39, 1615-1621
Halland, N., Braunton, A., Bachmann, S. et al. (2004). “Direct organocatalytic asymmetric alpha-chlorination of aldehydes.”Journal of American Chemical Society, 126, 4790–4791
Hayashi, Y., Yamaguchi, J., Hibino, K. et al. (2003). “Direct proline catalyzed asymmetric α-aminooxylation of aldehydes.” Tetrahedron Letters, 44, 8293– 8296
Hoang, L., Bahmanyar, S. and Houk, K. (2003). “Kinetic and stereochemical evidence for the involvement of only one proline molecule in the transition states of proline-catalyzed intra- and intermolecular aldol reactions.” Journal of American Chemical Society, 125, 16–17
Hoffmann, S., Nicoletti, M. and List, B. (2006). “Catalytic asymmetric reductive amination of aldehydes via dynamic kinetic resolution.” Journal of American Chemical Society, 128, 13074–13075
[Jacobsen, E. and Joly, G. (2004). “Thiourea-Catalyzed Enantioselective Hydrophosphonylation of Imines: Practical Access to Enantiomerically Enriched α-Amino Phosphonic Acids.” Journal of American Chemical Society, 126 (13), 4102–4103.
Jacobsen, E. N. (2002). “Asymmetric Catalysis of Epoxide Ring-Opening Reactions,” Acc. … for the Strecker Reaction,” Journal of American Chemical Society, 124, 10012–10014
Jen, W., Wiener, J. and MacMillan, D. (2000). “New strategies for organic catalysis: The first enantioselective organocatalytic 1,3-dipolar cycloaddition.” Journal of American Chemical Society, 122, 9874–9875
Jencks, W. (1969) Catalysis in Chemistry and Enzymology. New York: McGraw-Hill.
Kumaragurubaran, N., Juhl, K., Zhuang, Wet al. (2002). “Direct L-proline-catalyzed asymmetric alpha-amination of ketones.” Journal of American Chemical Society, 124, 6254–6255
Kunz, R. and MacMillan, D. (2005). “Enantioselective organocatalytic cyclopropanations. The identification of a new class of iminium catalyst based upon directed electrostatic activation.” Journal of American Chemical Society, 127, 3240–3241
Lacour, J. and Hebbe-Viton, V. (2003). “Recent developments in chiral anion mediated asymmetric chemistry.” Chemical Society Review, 32, 373–38
Li, X. and List, B. (2007). “Catalytic asymmetric hydrogenation of aldehydes.” Chemical Community, 17, 1739–1741
List, B (2002). “Proline-catalyzed asymmetric reactions.” Tetrahedron, 58:5573– 5590
List, B and Yang, J. (2006). “Chemistry. The organic approach to asymmetric catalysis.” Science, 313, 1584–1586
List, B. (2000a). “The direct catalytic asymmetric three-component Mannich reaction.” Journal of American Chemical Society, 122, 9336–9337
List, B. (2001b). “Asymmetric aminocatalysis.” Synletters, 2001, 1675–1686
List, B. and Hechavarria, F. (2004). “Catalytic asymmetric intramolecular Michael reaction of aldehydes.” Angewandte Chemie International Edition, 43, 3958–3960
List, B., Lerner, R. and Barbas, C. (2000b). “Proline-catalyzed direct asymmetric aldol reactions.” Journal of American Chemical Society, 122, 2395–2396
March, J. (1992) Advanced Organic Chemistry. New York: Wiley-Interscience
Masel, I. (2001) Chemical Kinetics and Catalysis. New York: Wiley-Interscience.
Mikaki, K. and Lauterns, M. (2007) New Frontiers in Asymmetric Catalysis. New York: Wiley.
Nicolaou, K. and Sorensen E. (1996) Classics in total synthesis. Weinheim: Wiley-VCH.
Nishiyama, H. and Itoh, K. (2000) Asymmetric hydrosilylation and related reactions. In: Ojima I (eds) Catalytic asymmetric synthesis. New York: Wiley- VCH.
Northrup, A. and MacMillan, D. (2002). “The first direct and enantioselective cross-aldol reaction of aldehydes.” Journal of American Chemical Society, 124, 6798–6799
Noyori R (2002) Asymmetric catalysis: science and opportunities (Nobel lecture). Angewandte Chemie International Edition, 41:2008–2022
Noyori, R. Tokunaga, M. and Kitamura, M (1995). “Stereoselective organic synthesis via dynamic kinetic resolution.” Bull Chemical Society of Japan, 68, 36–55
Schinzer, D. (eds.). (1989). Selectivities in Lewis Acid Promoted Reactions. Dordrecht: Kluwer Academic Publishers.
Schreiner, P. (2003). “Metal-free organocatalysis through explicit hydrogen bonding Interactions.” Chemical Society Review, 32, 289–296
Seayad, J. and List, B. (2005). “Asymmetric organocatalysis.” Organic Biomolecular Chemicals, 3, 719– 724
Sevin, A., Maddaluno, J. and Agami, C. (1987). “Compact transition states and asymmetric induction: a theoretical ab initio SCF-CI study of enamine addition to carbonyl compounds. Journal of Organic Chemistry, 52, 5611-5615
Tang, Z., Luo-Ting, F. J., Cui, X. et al. (2003). “Novel Small Organic Molecules for a Highly Enantioselective Direct Aldol Reaction.” Journal of American Chemical Society. 125, 5262-5263.
Tsuzuki, S., Honda, K., Uchimaru, T. et al. (2000) “The Magnitude of the CH/ð Interaction between Benzene and Some Model Hydrocarbons.” Journal of American Chemical Society, 122, 3746-3753.
Uraguchi, D. and Terada, M. (2004). “Chiral Brønsted acid-catalyzed direct Mannich reactions via electrophilic activation.” Journal of American Chemical Society, 126, 5356–5357
Ward, R. (1995). “Dynamic kinetic resolution.” Tetrahedron Asymmetry, 6, 1475–1490
Wilson, R., Jen, W. and MacMillan, D. (2005). “Enantioselective organocatalytic intramolecular Diels–Alder reactions. The asymmetric synthesis of solanapyrone.” Journal of American Chemical Society, 127, 11616–11617
Yamamoto, H. (Eds.), (2000) Lewis Acids in Organic Synthesis. Weinheim: Wiley-VCH.