Abstract
Sickle cell disease (SCD) is an inherited genetic disorder that arises from a single nucleotide polymorphism in the beta-globin gene. The resultant change of amino acid in the globin part affects the stability of the protein, which leads to the polymerization of two beta-globin chains of adjacent molecules. The new structure distorts the shape of the red blood cells and affects their flexibility thereby leading to the blockage of blood vessels in what is called vaso-occlusion. Intensive pain, which arises from vaso-occlusion and acute chest syndrome, is one of the complaints of sickle cell disease patients. The extent of the disease is influenced by the type of hemoglobin inherited from a child’s parents. The diagnosis of SCD is performed when a child is born and involves techniques such as high-performance liquid chromatography, solubility tests, gel electrophoresis and isoelectric focusing. The treatment of sickle-cell disease centers on managing the complications associated with the disease such as renal loss, acute chest syndrome, acute vaso-occlusive pain, bacterial infections, and heart disorders, pulmonary hypertension, and neurological conditions. However, some of the available treatment options pose some shortcomings. Therefore, there is a need to carry out intensive research to develop advanced treatment options. Possible areas for future research include gene therapy and stem-cell research.
Introduction
Sickle cell disease (SCD) is a severe inherent genetic disease that affects approximately 100,000 people in the U.S. (Roseff 2009). The disorder arises when there is a transmutation in the beta-globin genes, which are responsible for the formation of functional hemoglobin, the major protein that facilitates the transport of oxygen in the body. Therefore, the disorder is associated with a number of critical and lingering problems that require prompt medical intervention. SCD affects several body systems and is known to cause incidents of severe illness and gradual organ destruction. Consequently, it is among the most prevalent life-threatening monogenic disorders globally.
The understanding of the disease over the years has contributed to the knowledge base that has facilitated the development of intervention strategies. For example, the typical sickle-shape of erythrocytes in SCD was first elucidated in 1910 by a biologist called Herrick (Rees, William, & Gladwin 2010). Thereafter, in 1949, other scientists recognized the electrophoretic irregularities in sickle hemoglobin, which is denoted as HbS and created the term “molecular disease.”
Hemoglobin comprises an iron part (heme) and four protein (globin) chains. The attributes of the globin chains control the type of hemoglobin as indicated in Table 1.
Table 1: The constituents of normal adult hemoglobin.
SCD is marked by the presence of anomalous hemoglobin known as hemoglobin S, the disorder is described as a hemoglobinopathy. The disorder manifests when a child inherits faulty hemoglobin genes from both parents. The hemoglobin constitution for patients with homozygous SS is shown in Table 2.
Table 2: The constituents of abnormal hemoglobin.
Other forms of aberrant hemoglobin may be found in the presence or absence of Hb S. The type of hemoglobin possessed by an individual is determined by the pattern of inheritance. The contribution of hemoglobin S by each parent leads to the inheritance of two copies of Hb S and is indicated as Hb SS (homozygous for hemoglobin S). Conversely, inheriting only one copy of the defective hemoglobin from one parent and a normal copy of hemoglobin gives rise to a heterozygous individual described as Hb AS. Such individuals do not display any symptoms associated with the disease except under exceptional conditions where they present with hypoxia, particularly at elevated altitudes. Hence, not everyone with defective hemoglobin develops the disease. Even though Hb S is found universally, it is prevalent in Western Africa (Quimby et al. 2014).
The identification of SCD is done following neonate screening. Previously, young children had signs such as agonizing protuberances of the limbs, which were a result of vaso-occlusion. In most SCD patients, there is anemia with hemoglobin levels ranging from 6 g/dL to 8 g/dL. Their peripheral blood smears show distinctive sickle-shaped cells as shown in Figure 1.
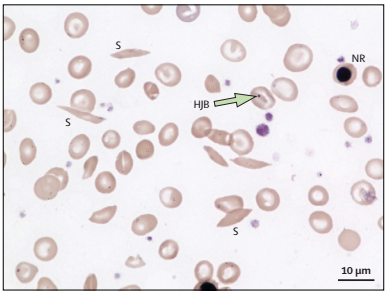
However, SCD patients do not always have suggestive anemia because the body recompenses the peripheral red blood cell (RBC) damage by elevated erythropoiesis in the bone marrow. Thus, the bone marrow holds many RBC precursors, which leads to the untimely release of undeveloped precursors (reticulocytes) from the marrow. Peripheral smears also reveal the under-developed RBCs, which are usually bigger than fully-developed erythrocytes RBCs and have a mild blue coloration due to incomplete hemoglobinization.
The biophysics of hemoglobin and the heredities causing the disorder have been investigated widely and have improved the comprehension of other molecular maladies. Previously, children diagnosed with SCD did not live to celebrate their 20th birthdays. Nevertheless, advances in the medical management of sickle-cell disease have reversed this trend. Even though the available treatment options do not address the pathophysiology of the disease, there is hope that better interventions strategies will be made available. The aim of this paper is to illustrate the pathophysiology of SCD, the current strategies used in its management and novel areas in SCD therapeutics.
Pathophysiology of Sickle-Cell Disease
HbS arises from the change in the β-globin gene, which is a single nucleotide polymorphism that alters the 17th nucleotide from thymine to adenine. Consequently, the sixth amino acid in the resultant protein becomes valine as a replacement for glutamic acid. This change generates a hydrophobic pattern in the deoxygenated HbS tetramer, which causes the linkage of the β1 and β2 chains of separate hemoglobin molecules. This linkage is referred to as crystallization and yields a polymer nucleus that occupies space in the erythrocyte thereby interfering with its build and elasticity. Crystallization also favors cellular dehydration alongside physical and oxidative stress to the cell. The degree of polymerization of HbS is relative to the level and length of hemoglobin deoxygenation, the amount of intracellular HbS, and the existence of fetal hemoglobin. Fetal hemoglobin substantially lowers the amount of HbS.
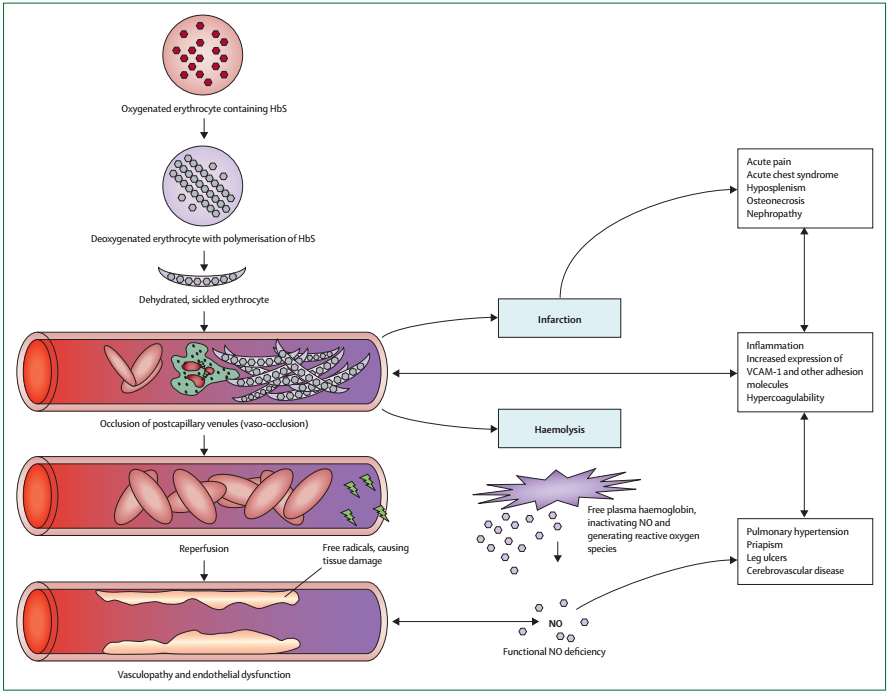
The severity of the disease is influenced by the extent and progression of HbS polymerization, which is demonstrated by simultaneous inheritance of hereditary attributes that control the concentrations of HbS or fetal Hb. Two main pathophysiological processes namely vaso-occlusion with ischemia-reperfusion injury and hemolytic anemia occur in the course of the disease.
Intense vaso-occlusive pain is brought about by the capturing of RBCs and white blood cells (WBCs or erythrocytes) in the capillaries, which blocks the blood vessels causes tissue ischemia. Inflammatory processes have been reported to set off vascular barricades by sickle erythrocytes. This idea has been demonstrated by the induction of endothelial leucocyte-erythrocyte binding connections in HbS mice treated with inflammatory drugs (Quimby et al. 2014). Apart from inflammatory triggers, capillary obstruction by rigid, deformed erythrocytes with high HbS polymer content also contribute to microvascular vaso-occlusion.
Vascular occlusion, conversely, is a consequence of the active associations between RBCs and the endothelium of the blood vessels leading to sporadic microvascular obstruction and ischemia, followed by reinstatement of blood movement, which causes additional damage to tissues. The ischemia and reperfusion progressions produce oxidative stress, which triggers vascular oxidases and fiery stress. The number of endothelial cell adhesion molecules increases as well as inflammatory cytokines, which work together to instigate leukocytosis. The lack of oxygen in the bone marrow generates fat residues that block blood vessels, particularly in the lungs leading to acute chest syndrome.
Hemolytic anemia is another pathological process that results from HbS polymerization. Previous studies show that hemolysis promotes anemia, exhaustion and gall bladder stones. Recent investigations show that it also triggers the advancement of progressive vasculopathy. The risk of vasculopathy marked by elevated blood pressure, endothelial malfunction, and proliferative alterations of walls of blood vessels increases with advancing age. Figure 2 summarizes the pathophysiology of sickle-cell disease.
Diagnostic and Screening
Most states require newborns to be screened for various illnesses following birth. These screenings enable the identification of babies with SCD. Some of the diagnostic methods used include High-performance liquid chromatography (HPLC), hemoglobin electrophoresis, isoelectric focusing, and solubility tests. HPLC is regarded among the most accurate techniques of recognizing aberrant hemoglobin forms and setting them apart from normal hemoglobin. HPLC can also determine the concentrations of various types of hemoglobin. Hemoglobin electrophoresis segregates various hemoglobins based on their archetypal passage across an electrophoretic gel. The commonly used gels are cellulose acetate at basic pH or acidic citrate agar. Therefore, the motion of the hemoglobins on the gel depends on their net charge and causes them to occupy distinct positions on the gel. Isoelectric focusing utilizes an electric current to cause further separation of hemoglobin on the electrophoretic gel. The identity of the hemoglobin is made from the observed pattern of migration. Solubility tests manipulate the insolubility of hemoglobin S in certain solutions. Therefore, an uncomplicated solubility test can enable the detection of Hb S. The erythrocytes are first lysed before being introduced to a solution of sodium hydrosulfite, which reduces them. The presence of Hg S is indicated by the turbidity of the mixture. The shortcoming of this test is that it requires the use of a specified of whole blood and is unsuitable for neonates. Another shortcoming is that individuals who are heterozygous for HbS will test positive for the disorder.
Management of Sickle-Cell Disease
The management of sickle cell disease focusses on alleviating the presenting symptoms and providing patient comfort. Some of the complications associated with sickle cell disease include acute pain, infections, neurological complications, acute chest syndrome, heart disease, pulmonary hypertension, and kidney problems. Therefore, the treatment of SCD addresses each complication specifically.
Most hospital admission in SCD patients including adults is due to acute vaso-occlusive pain. Even though acute vaso-occlusive agony resolves itself and does not result in irreversible organ damage, it is the most significant complication from the patient’s point of view. Immense pain is associated with SCD mortalities in patients above the age of 20 years. Opiate analgesics are mostly used to alleviate severe pain. Data from private investigations on the deaths of SCD patients reveal opiate-related over-sedation, which emphasizes the need to keep an eye on patients on opiate analgesics (Jordan, Swerdlow, & Coates 2013).
Bacterial infections are implicated in the deaths of children with SCD, who are more susceptible to bacterial infections due to diminished splenic function, flaws in complement activation, inadequate micronutrients, and tissue ischemia. The most notable infections are Streptococcus pneumonia, Haemophilus influenza, and non-Typhi Salmonella species (Yordan et al. 2014). The introduction of penicillin prophylaxis and vaccination with conjugate vaccines against these microbes has led to significant improvements in prognosis.
Neurological snags such as strokes are common in SCD children. These disorders are attributed to vasculopathy that interferes with the carotid and cerebral arteries. According to the Stroke Prevention in Sickle Cell Anemia (STOP) research, the most effective way of reducing the chances of stroke is to provide frequent blood transfusions. The transfusions reduce the concentration of Hb S and lower the risk of stroke by 90% (Lee et al. 2006). A relatively new program of transcranial Doppler screening has also been reported to reduce the risk of stroke. Treatment with hydroxyurea yielded disappointing outcomes in clinical trials.
Acute chest syndrome also leads to frequent hospital admissions. Its treatment involves the administration of broad-spectrum antibiotics, bronchodilators, and oxygen. Blood transfusions are performed in cases of very low hemoglobin levels. The prompt use of top-up transfusion is preferred to exchange transfusion, which is often used as a last resort in isolated cases. Corticosteroids such as dexamethasone lead to improved outcomes and eliminate the need for transfusions. However, their use is restricted due to the incidence of recoiling pain once the corticosteroids are discontinued.
Pulmonary hypertension, which may be denoted as pulmonary artery systolic pressure surpassing 35 mmHg, is common in SCD patients and increases the risk of mortality. Evidence regarding the best ways of managing pulmonary hypertension in SCD patients is still lacking. Therefore, the risk factors of the complication such as hypoxemia, restrictive lung disorder, sleep apnea, severe anemia, and iron burden among others need to be recognized and managed. Some of the currently used treatment options include hydroxycarbamide and frequent blood transfusions. Catheterization may be done to evaluate the left ventricular function in some instances.
Diastolic malfunction often causes left-sided heart disorder in approximately 10% of SCD patients and is an autonomous risk factor for death. The presence of pulmonary vascular disorder and diastolic malfunction increases the risk of death significantly. Pulmonary pressures increase dangerously during vaso-occlusive pain acute chest syndrome. Therefore, managing the causative conditions help to reduce the chances of heart disease.
Renal loss is to be expected in sickle-cell disease due to the increased likelihood of Hb S polymerizing in the renal medulla. RBC dehydration then ensues from the decreased partial pressure of oxygen, acidic pH and elevated osmolality. The resultant vaso-occlusion leads to the death of renal tissues marked by papillary necrosis and scarring of the medulla. The treatment of renal disorders in SCD entails the prompt initiation of hydroxycarbamide and angiotensin-converting enzyme inhibitors in patients with substantial albuminuria. Renal transplants have also produced encouraging results.
Future Developments
The improvement of SCD therapy is likely to take advantage of novel technologies such as stem-cell transplantation and gene therapy. Studies on the use of induced pluripotent stem cells in managing genetic disorders have yielded encouraging findings (Ye et al. 2009). Information from genetic correlation studies is likely to uncover more epigenetic factors to elucidate the phenotypic diversity in SCD thus facilitating better prognosis and proper management of the specific problems experienced in SCD. Several pharmaceutical approaches have proven useful in vitro without demonstrating clinical usefulness. Some of these options include the use of heparin and sulfasalazine, which act as anticoagulants, to minimize the triggering of the endothelial and prevent the adhesion of RBCs. Another viable option to reduce inflammation is the use of corticosteroids such as dexamethasone and secretory phospholipase A2 blockers such as varespladib (Rees, William, & Gladwin, 2010). Therefore, future studies should look into these drugs to identify efficacy gaps and develop better treatments for sickle cell disease. Additionally, there is limited information regarding sickle cell disease in Africa, which has been reported to have high incidences of SCD (Modell & Darlison 2008). Future studies should also find ways to improve the knowledge of SCD in Africa and other regions of the world to facilitate improved management of patients globally.
Conclusions
Sickle-cell disease is a heritable disorder that affects a large number of people and was previously associated with early death. However, intensive research has led to improved strategies for managing the disease, which has improved the life expectancy of SCD patients. From the findings of this paper, it can be concluded that a wealth of research has tremendously improved the management of sickle-cell disease leading to improved outcomes. However, various gaps still exist, for example, evidence-based ways of managing some of the complications of SCD such as heart disease and hypertension. Some of the drugs used such as corticosteroids have been shown to be effective albeit with adverse effects such as recurrence of the condition once the drug is discontinued. Therefore, future studies need to focus on other possible pharmacologic and non-pharmacologic interventions to improve the prognosis of the disease.
References
Jordan, L., Swerdlow, P. and Coates, T. D. 2013. A systematic review of the transition from adolescent to adult care in patients with sickle cell disease. J. Pediatr. Hematol. Oncol 35:165-169.
Lee, M.T., Piomelli, S., Granger, S., Miller, S.T., Harkness, S., Brambilla, D.J. & Adams, R. J. 2006. Stroke Prevention Trial in Sickle Cell Anemia (STOP): extended follow-up and final results. Blood 108:847-852.
Modell, B. and Darlison, M. 2008. Global epidemiology of hemoglobin disorders and derived service indicators. Bull. World Health Organ 816:480-487.
Quimby, K.R., Moe, S., Sealy, I., Nicholls, C., Hambleton, I.R. and Landis, R.C. 2014. Clinical findings associated with homozygous sickle cell disease in the Barbadian population–do we need a national SCD registry? BMC Res. Notes 7:102.
Rees, D. C., William, T. N., and Gladwin, M. T. 2010. Sickle-cell disease. Lancet 376:2018-20131.
Roseff, S. D. 2009. Sickle cell disease: a review. Immunohematology 25:67-74.
Yawn, B.P., Buchanan, G.R., Afenyi-Annan, A.N., Ballas, S.K., Hassell, K.L., James, A.H., Jordan, L., Lanzkron, S.M., Lottenberg, R., Savage, W.J. and Tanabe, P.J. 2014. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 312:1033-1048.
Ye, L., Chang, J.C., Lin, C., Sun, X., Yu, J. and Kan, Y.W. 2009. Induced pluripotent stem cells offer a new approach to therapy in thalassemia and sickle cell anemia and option in prenatal diagnosis in genetic diseases. Proc. Natl. Acad. Sci 106:9826-9830.