Abstract
Many malignancies depend on unique body processes such as angiogenesis for their progression and survival. Management of malignancies entails targeting specific components behind the processes such as the vascular endothelial growth factor (VEGF) and/or associated receptors. This study investigates the outcome of viral delivery systems like soluble vascular endothelial growth factor receptor Fms-like tyrosine kinase-1 (sFLT-1) in acute myeloid cells. A recombinant lentiviral vector expressing sFLT-1 was constructed and produced through co-transfecting the pLenti-sFLT-1 and pLenti6.3/V5-GW/EmGFP) into the 293FT Cell Line. Western blotting was performed to identify the VEGF165 protein in these cells. Amplification via reverse transcription-polymerase chain reaction of sFLT-1 cDNA was then carried out. Inhibitory effect of sFLT-1 on K562 and HL60 cells proliferation was assessed and subsequent optimizations of LV-sFLT-1 copy number carried out by enzyme-linked Immunosorbent assay (ELISA). Treatment with Lenti-sFLT-1 had a significant inhibitory impact on the proliferation of acute myeloid cells. Our results imply that lentivirus-mediated sFLT-1 gene therapy is effective and can provide new frontiers in the therapy of AML.
Introduction
Acute Myeloid Leukemia (AML) is the unrestricted and rapid proliferation of myeloid cells due to mutations in the progenitor hematopoietic cell line. Age is a key factor differentiating molecular pathogenesis of AML with children having more chromosomal translocations and adult pathogenesis entails AML chromosomal losses and gains (Grimwade, 2001). These mutations and translocations in signal transduction genes generate chimeric fusion proteins which are the main pathogenic components in AML (Kelly & Gilliland, 2002). Current treatment modalities of AML such as immunotherapeutic approaches suffer from a myriad of setbacks such as the difficulty in finding a compatible sibling donor (Kamps et al, 2010). In addition, pharmacological approaches have encountered difficulties such as resistance, toxicity, and incomplete remission of the tumor (Kerbel, 2001).
Angiogenesis has been postulated to play a role in pathogenesis in AML. Angiogenesis is a product of the balance between pro-angiogenic and antiangiogenic growth factors and cytokines (Ribatti, Vacca &Presta, 2000; Hussong, Rodgers & Shami, 2000). Research in the pathogenesis of AML has identified vascular endothelial growth factor (VEGF), to be among the most specific and essential regulators of the process of angiogenesis. VEGF regulates angiogenesis by modulating processes such as endothelial proliferation, permeability and survival. VEGF consists of five family factors of which VEGF-A is most studied and understood such that it is usually referred to as VEGF. Expression of VEGF-A is the form of foyur characterized isoforms namely 121, 165, 189, and 206. Of these isoforms of VEGF-A, isoform 165 is the principal isoform found to be expressed in AML tumor cells in both adults and children (Hicklin DJ, Ellis LM. 2005 Keyt BA, Berleau LT, Nguyen HV, et al. 1996). The role of VEGF in angiogenesis is mediated via specific tyrosine kinase receptors occurring on the endothelial cells (List, 1999).
Recent studies show that acute myeloid cells express a rich presence of VEGF as well as its receptors which are implicated in facilitation of progression and survival of acute myeloid leukemic progenitors (Frankel & Gill, 2004). Studies show that VEGF mediated autocrine and paracrine signals are involved in infiltration of leukemic cells in bone marrow, liver, and spleen as well as increased microvascular density in bone marrow of AML patients (Aguayo et al, 1999; Schuch, 2002; Padro et al, 2000). In addition, evidence brought forth show a linear correlation of the expression of VEGF and its receptors by AML blasts with reported low rates remission and overall survival among AML patients (Padro et al, 2000). Conversely, it was reported that VEGF has the ability to inhibit apoptotic death in hematopoietic cells following exposure to
Pharmacologic agents and as such, expression of VEGF and its receptors by leukemic cells not only is implicated in prolongation of survival of leukemic cells but also is involved in promotion of sensitivity of leukemic cells for pharmacologic agents (Kamps et al, 2001).
The application of Gene therapy in conjunction with other treatment modalities provides a promising novel approach in management of numerous hematological malignancies as it provides a means of circumventing setbacks suffered by current treatment modalities. The dual approach may result in improving the response by patients to already available conventional treatment (Grimwade, 2001; Kelly & Gilliland, 2002; Kume, Hanazono, Mizukami, Okada, & Ozawaa, 2002). Inhibitors of VEGF such as the sFLT-1 hold a lot of promise on approach in regulation of angiogenesis a major factor implicated in growth, development and survival of many hematological malignancies including AML. sFLT-1, an endogenously expressed entity, binds with very high affinity to VEGF without initiation of signal transduction and as such, results in site specific inhibition of the function of VEGF leading to regulation of angiogenic process in acute myeloid cells. Even though gene therapy modalities have been designed, the production and utilization of a number of angiogenic factors innate to the body has presented a problem.
In this study, we investigated the effects of sFLT-1 gene delivery on acute myeloid leukemic cells. We constructed the sFLT-1 gene that codes the 1–3 immunoglobulins (Ig)-like domains and established a lentiviral vector expressing sFLT-1. We investigated delivery of LV-sFLT-1 in acute myeloid leukemic cells and VEGF-mediated inhibitory effects of sFLT-1 in vitro.
Methodology
Cell culture
HL-60 (pro-myelomonocytic), BV173 (human, peripheral blood, leukemia, pre-B cell) K562 (myelogenous leukemia, erythroleukemia type), and CHO (Chinese hamster ovary) cells were grown in RPMI. Human umbilical vein endothelial cells (HUVEC) obtained from American Type Culture Collection (ATCC) were maintained in F12K supplemented with endothelial cell growth supplement (ECGS) (BD Biosciences) and heparin (Sigma). MCF7 cells were cultured in Dulbecco’s modified Eagles’s medium (DMEM); 293FT (human embryonal kidney) cells (Invitrogen) were cultured in DMEM supplemented with 200 mM L-Glutamine (Mediatech), 10 mM MEM non-essential amino acids (Gibco), 100 mM MEM sodium pyruvate and 10% fetal bovine serum (Hyclone Laboratories, Logan, UT). 293FT cells were used within 16 passages.
With the exception of 293FT cells, the media for cells were supplemented with penicillin, streptomycin (P4458, Sigma, USA), amphotericin B (A6804, Sigma, USA), bicarbonate (90421C, Sigma, USA) and 10% FBS (HyClone, Austria). Cells were obtained from American Type Culture Collection (ATTC). All cells were incubated in a CO2 incubator (Galaxy, UK) under 5% CO2 with more than 90% humidity. Subcultures were carried out when cells were confluent.
Identification of VEGF165 and VEGF R-1 expression in K562 and HL60 cells
Detection of VEGF165 and VEGF R-1 expression in our leukemic cells at the mRNA level, was carried out prior to investigation of the effects of sFLT-1 expression from the recombinant plasmids To achieve this we performed RT-PCR for the amplification of VEGF165 and VEGF R-1 in K562 and HL60 cells and also Western blotting to identify the VEGF165 protein.
MasterPure RNA Purification Kit (EPICENTRE) was used in the isolation total RNA from K562 and HL60 leukemic cells which was then used in synthesis of cDNA using the RevertAid™ H Minus First Strand cDNA Synthesis Kit and random hexamer primer (Fermentas, Lithuanian). Amplification of VEGF165 and VEGF R-1 were carried out using cDNA. RT-PCR reactions were carried out with the following primers: VEGF165: sense primer 5’-ACCCTGATGAGATCGA-GTAC-3’anti-senseprimer5’-TAATCTGCATGGTGATG TTGG – 3’, VEGF R-1: Sense Primer 5’- AC TCTA
ACATGCACCTGTGTGG- 3’ anti-sense Primer 5’- GCTCATCC- AAAGGA
ACT TCATCTG- 3’, Human GAPDH: Sense primer 5’- ACA ACAGCTCAAGATCATCAG-3’ anti-sense primer 5’- ATG AGT CCT TCC ACGA TACC-3’ with the expected sizes of 131, 145 and 121 bp respectively.
The PCR reaction consisted of a series of timed temperature treatments to obtain the PCR products. The bands were visualized using UV light and a molecular weight DNA ladder marker was used as a reference.
K562 and HL60 cells were collected in an appropriate centrifuge tube and centrifuged. The cells were rinsed once with PBS to remove residual medium. ProteoJET™ Mammalian Cell Lysis Reagent was added to packed cells and resuspended by vortexing and incubated. The lysates were clarified by another centrifugation process. The supernatants were transferred to new tubes. The cell lysates were immediately used or stored at -80°C for later use.
Protein quantification was performed for the extracted protein samples from K562 and HL60 cells in the same way as before, and the samples were used for Western blotting.
Western blotting was carried out by adding the appropriate primary antibody (Anti Mouse-VEGF165 1/250 Santa Cruz USA) and Mouse monoclonal ßactin (Ab1/5000 Abcam USA), in TBS for immunoprecipitation onto the membrane and was shaken at 4°C overnight. The membrane was then washed three times with TBST for 10 minutes each wash to remove unbound antibodies. After washing, alkaline phosphatase-conjugated secondary antibody Goat anti-mouse Ab 1/5000 (Chemicon, USA) in TBS was added to react with the primary antibody and left to shake for another 1 h at RT. The membrane was further washed three times with TBST solution, and lastly, BCIP(33 ul)/NBT(66 μl) solution in alkaline phosphatase buffer (100 mM Tris, 100 mM NaCl, 5 mM MgCl2; pH 9.5) was added as the substrate for the alkaline phosphatase. The reaction was stopped when the desired protein bands appeared. The membrane was washed thoroughly with water before being air-dried.
Cloning of the 1−3 Ig-like domains of FLT-1
RNA extraction and cDNA synthesis
The previous results regarding sFLT-1 effects on the myeloid leukemic cell lines presented no inhibitory effect of sFLT-1 in K562 and HL60 cell lines in both non-viral and viral delivery system. Therefore, to determine the VEGF mediated suppressory effect of sFLT-1 on leukemic cells at the mRNA level, we investigated VEGF mRNA levels in LV-sFLT-1 transduced K562 cells using Real Time PCR. The K562 cells were transduced with LV-sFLT-1 and LV-EmGFP in a 24-well cell culture plate. The cells were subjected to RNA extraction and cDNA synthesis, before, 24 and 48 h infection. To perform a quantitative Real Time PCR, standard curves were prepared for VEGF165 and a house-keeping gene (GAPDH). To prepare the standard curves, the BV173 cell line was chosen for VEGF165 amplification. Therefore, firstly RT-PCR was performed to detect VEGF165 in BV173 cells for optimization of amplification.
RNA extraction and cDNA synthesis were carried out. Then the synthesized cDNA (using random hexamer primer) was subjected to PCR for amplification of VEGF165. The primers used for PCR were designed and synthesized by Sigma (Singapore). VEGF165: sense primer 5’-ACCCTGATGAGA TCGAGTAC-3’anti-senseprimer 5’-TAATCTGCATGGTGATG TT GG- 3’, Human GAPDH: sense primer 5’- ACA ACAGCT CAA GATCATCAG-3’ anti-sense primer 5’- ATG AGT CCT TCC ACGA TACC-3’.
The bands were visualized using UV light and a DNA molecular weight ladder marker was used as a reference.
Real Time PCR was performed to detect the relative concentrations of VEGF mRNA in K562 cells transduced with LV-sFLT-1 and LV-EmGFP, with the untransduced cells as a control. Quantitative Real Time polymerase chain reaction was performed using the Real Time TaqMan PCR technique. Real Time PCR products were quantified relative to an endogenous control gene (GAPDH) to normalize quantification of target genes for differences in the amount of total nucleic acid added to each reaction. The control cell line (BV173) was used to construct standard curve. The concentration of BV173 cDNA was measured by Nanodrop (Thermo, USA) and the standard cDNA (template) was serially diluted in nuclease-free water by ten-fold dilutions of a cDNA. The PCR reactions were prepared by assembling Sigma’s reference dye, MgCl2 (1.7mM), cDNA template from each dilution, primers of Taq Man probe, and of 2X JumpStart Taq ReadyMixä with dUTP (Sigma Aldrich), topping up to a final volume in PCR tube. A nontemplate control (NTC) was prepared for amplification as well. The reaction was set manually in a 36-well rotor (Rotor-Gene 6000, Corbett, and Sydney, Australia). All reactions were prepared in triplicates. The cycling program for all standards were: 40 cycles of amplification, at 94°C for 2 minutes, 94°C for 15 seconds, annealing at 55°C for 40 seconds, and extension at 72°C for 45 minutes, the gain was set at 5.33 in green channel in Real Time PCR cycler Rotor–Gene 6000 (Corbett Life Science, Australia). The standard curves were obtained for VEGF165 and GAPDH by plotting the threshold cycle (Ct) on the Y-axis and the log of concentration (ng/μl) on the X-axis. Ct, slope, PCR efficiency, correlation co-efficient (R2) and percentage of variance in ng/μl were calculated by using the default settings of Rotor-Gene 6000. The same fluorescence thresholds used to construct the standard curves were also used to determine the Ct values of samples. The Ct values and concentration of samples (ng/μl) were calculated using the Rotor-Gene 6000 software and the samples were normalized to the endogenous control (GAPDH). The normalized samples signals was divided by the normalized control sample signal to determine the expression ratio of VEGF165 in transduced cells using 2 Std Curves. Diluted standards were stored at -80oC for the next experiments. The products were then separated by electrophoresis on 1% agarose gel and the bands were visualized using UV light (Alpha–Innotech gel documentation system) to check the appropriate size of products in comparison with the appropriate DNA ladder Marker. The attB-primers were used for amplification of the attB-sFLT-1 PCR products. The entry clones generated by BP recombination were sequenced using
M13 sequencing primers (M13 Forward (-20) priming site: GTAAAACGACGGCCAG, M13 Reverse priming site: GTCATAGCTGTTTCCTG) by NHK Bio-Science, Korea).
Construction of a recombinant lentiviral vector expressing sFLT-1
The sFLT-1 gene was recombined into the pLenti6.3/V5–DEST GatewayR Vector to facilitate expression of the gene of interest. The pLenti-DEST vectors are unique vectors adapted to be utilized by the Gateway® Technology. They are made in such a way as to permit complex expression of recombinant fusion proteins in living cells (Boshart, 1985).
Production of the sFLT-1 PCR product containing attB sites was carried out before the sFLT-1 gene was cloned into a GatewayR entry vector (BP Recombination Reaction). After that, an expression clone was produced by performing an LR recombination reaction between the entry clone and a GatewayR destination vector (pLenti 6.3/V5–DEST Invitrogen). And lastly, the expression clone was used in the ViraPower™ HiPerform™ Lentiviral Expression System (Invitrogen).
Generation of Recombinant lentivirus-expressing sFLT-1 and GFP) LV-sFLT-1 and LV- EmGFP)
Lentivirus production was performed by co-transfecting the optimized packaging plasmid mix and the pLenti expression constructs (pLenti-sFLT-1and pLenti6.3/V5-GW/EmGFP) (invitrogen) into the 293FT Cell Line (Invitrogen). The co-transfection of 293FT cells was performed using the reverse transfection procedure. The obtained LV-sFLT-1 was then Concentrated. The 293FT cells were transduced to titer the efficiency of lentivirus expressing GFP (Invitrogen) in the presence of hexadimethrine bromide (Polybrene®, Sigma, Catalog no. H9268). The titer of Lentiviral expressing EmGFP was detected by fluorescent detection of EmGFP using a flow cytometer and expressed in transducing units (TU)/ml. Estimation of the intensity of GFP in transduced cells at 48 and 72 h post-infection was then carried out. Thereafter, Titering Lentivirus-sFLT-1 (LV-sFLT-1) was done and a standard curve was obtained for sFLT-1 by plotting the threshold cycle (Ct) on the Y-axis and the log of concentration (ng/μl) on the X-axis. Ct, slope, PCR efficiency, correlation co-efficient (R2) and percentage of variance in ng/μl were calculated. The same fluorescence thresholds used to construct the standard curves were also used to determine Ct values. The specificity of the reaction was shown by the detection of the Tms of the amplification products directly after the last reaction cycle. A post-PCR process was analyzed to obtain the melt curve of PCR product. A standard curve was then plotted for the calculation of PCR efficiency and quantification.
Verification of expression of sFLT-1 by enzyme-linkedimmunosorbent assay (ELISA), RT-PCR and western blotting
To detect an optimal amount of LV-sFLT-1 to express the highest level of sFLT-1 protein in transduced cells, the K562 cells seeded in 12-well cell culture plate (2 x105) were transduced with different copy numbers of LVsFLT-1. The copy numbers of LV-sFLT-1 in 10-2, which was determined by Real time PCR, was considered as a base and the desired copy numbers were counted from that in a 500μl infection mediasFLT-1 protein expression was confirmed and quantified by performing a quantitative sandwich enzyme immunoassay technique, using human soluble VEGF R-1 /FLT-1 ELISA kit. For this experiment, the cell supernatant from each well was collected and centrifuged at 1000 x g for 5 minutes, followed by aliquoting in 1.5 ml tubes and storage in –80°C. The ELISA assay was performed according to the protocol of the kit. The wash buffer dilution, VEGF R-1 standard reconstitution and preparation of standard curve dilutions by two-fold dilution was carried out and 100μl of assay diluents RD1-68 was added to each well. Then, 100μl of standards, controls, and samples were added (in triplicates) and mixed by incubating the plates at RT on a horizontal orbital microplate shaker at 300 rpm for 3 h. After incubation, washing was done for a total of four washes and 200μl of VEGF R-1 conjugate was added to each well and incubated for 3 h at room temperature on the shaker (300 rpm) followed by washing. 200μl of substrate solution was added to each well and incubated for 30 minutes at RT (protected from dark). The reaction was stopped by adding 50μl of stop solution to each well and the optical density of each well was determined, using a microplate ELISA reader at 450 nm with a wavelength correction of 570 nm. The concentration of samples was obtained by comparison with the standard curve.
K562 and HL60 cells proliferation inhibition assay
K562 and HL60 cells, at a density of 5×104 cells/well, were transduced with LV-sFLT-1(8 × 108 copies/μl) and LV-EmGFP in 24-well cell culture plates with 300μl infection media and incubated overnight in a cell culture incubator. Thereafter, the infection media was replaced with the complete growth media and incubated for 24 h. Then, the cells were incubated for 2 h in 500μl serum-free media and then 20ng/ml VEGF was added to the wells, followed by incubation for 72 h in a cell culture incubator. After 72 h, the K562 and HL60 cells were collected in 1.5 ml microcentrifuge tubes and centrifuged at 210 x g for 5 minutes. The cell pellets were resuspended in 200μl of growth media and 100μl of cell suspension transferred to 96-well cell culture plates following addition of 20μl MTS reagent to each well and incubated for 4 h at 37°C. The absorbance of formazan at 490 nm was determined on a multi-well plate reader.
HUVEC proliferation inhibition assay
The suppression of VEGF-driven proliferation of HUVECs by the sFLT-1 secreted in the conditioned media of transduced acute myeloid leukemic cells. the HUVECs were seeded in 24-well cell culture plates at a density of 1×105 cells/well containing 500μl growth media. 300μl conditioned media obtained from LV-sFLT-1 and LVEmGFP transduced K562 cells were added to the HUVECs, followed by removal of the growth media from the cells. After 15 minutes, 20ng/ml VEGF protein was added to each well and incubated at 37°C for 24 h. Then, 200μl of complete growth media was added to each well. The OptiMEM® I Medium was added for untreated control cells instead of conditioned media of transfected cells with the same condition. After 72 h, the proliferation of cells was estimated by MTS assay as described. The experiments were performed in triplicate.
HUVEC migration inhibition assay
The inhibitory effects of sFLT-1 expression from K562 cells in the migration of HUVECs were investigated using cell migration assay kit QCM 24-well Colorimetric Cell Migration assay utilizing an 8μm pore size, as this is appropriate for most cell types. According to the instruction of the kit, the HUVECs were cultured in 25 cm2 cell culture flasks at 70% confluency, containing conditioned media of LV-sFLT-1, LV-EmGFP transduced K562 cells, and OptiMEM® I Medium (70%) and growth media (30%). After 48 h, the cells were starved by incubation for 24 h prior experiment in serum-free media containing the conditioned media. Thereafter, the cells were harvested and a cell suspension containing 1 × 105 cells/ml was prepared.
300μl of cell suspension was added to each insert in triplicates. 500μl conditioned media of LV-sFLT-1 and LV-EmGFP transduced K562 cells containing 40ng/μl VEGF protein were added to the lower chamber incubated for 24 h at 37°C in aCO2 incubator. The cells/media were removed carefully from the top of inserts by pipetting out the remaining cell suspension, and the migration inserts were placed into a clean well containing 400μl of stain, followed by incubating for 20 minutes at RT. The inserts were rinsed by dipping inserts into water several times and cotton-tipped swabs were used to gently remove non migratory cell layers from the interior of the inserts. The inserts were allowed to dry and were transferred to the clean wells containing 200μl extraction buffer for 15 minutes at RT. Then, 100μl of the extraction mixture were transferred to a 96-well microtiter plate and the optical density measured at 560nm.
Results
Identification of VEGF165 and VEGF R-1 expression in K562 and HL60 cells
Cloning of the 1−3 Ig-like domains of FLT-1
In order to clone sFLT-1 into the pVAX1 vector, first the total RNA of HUVEC cells was extracted and qualified by a Nanodrop spectrophotometer (Figure 1). The cDNA was synthesized using the reverse sFLT-1 primer and quantified. The sFLT-1 cDNA was used for RT-PCR to amplify the sFLT-1 gene in gradient annealing temperatures of 65.3, 67.4 and 68.7°C (Figure 1). The pVAX1 vector was digested using the same restriction enzyme which was used for the amplification of sFLT-1 gene. Both digested vector and PCR product were subjected to gel purification and the purified products were confirmed in terms of purity and size by 1% agarose gel electrophoresis. The PCR product was ligated into double digested pVAX1. Then, the ligation mixture was transformed into E. coli, and positive recombinant plasmids were selected and subjected to single- and double-digestions to confirm the presence of the inserts (Figure 2). The construct was also sequenced to ensure the sequence accuracy and maintenance of the correct reading frame.
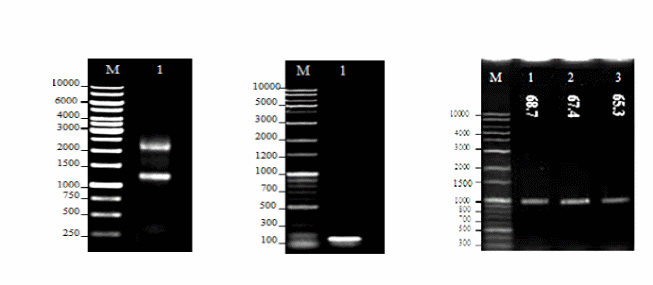
- (a) 1% Agarose gel electrophoresis of total RNA from HUVECs. Lane M: DNA marker (GeneRuler™ 1 kb DNA Ladder, 250-10000 bp, Fermentas, Lithuania); Lane 1: Total RNA using sFLT-1reverse primer, the 18S and 28S ribosomal RNA bands were observed.
- (b) 1% Agarose gel electrophoresis of amplified GAPDH from HUVEC cDNA. Lane M: DNA marker (Mbiotech’s 1Kb plus 100bp, Korea); Lane 1: Amplification of GAPDH (121bp).
- (c) 1% Agarose gel electrophoresis of amplified sFLT-1. RT-PCR was performed to amplify the sFLT-1 gene in a gradient of annealing temperatures. Lane M: DNA marker (Mbiotech’s 1Kb plus 100bp, Korea); Lane 1, 2, 3: Annealing temperatures of 65.3, 67.4, and 68.7°C, respectively. The amplified sFLT-1 was observed at all annealing temperatures at the size of interest (1000bp).

- (a) 1% Agarose gel electrophoresis of gel purified digested pVAX1 vector. Lane M: DNA marker (Mbiotech’s 1Kb plus 100bp, Korea); Lane 1: Undigested pVAX1; Lane 2: Single digested pVAX1 with Hind III; Lane 3: Double digested pVAX1 with Hind III and ECORI. Single and double- digested pVAX1 vector at the size of interest (3000 bp).
- (b) 1% Agarose gel electrophoresis of recombinant pVAX1-sFLT-1. Lane M: DNA marker (Mbiotech’s 1Kb plus 100bp, Korea); Lane 1: Single digested pVAX1 vector with Hind III (3000 bp); Lane 2: Single-digested pVAX1-sFLT-1 (4000 bp); Lane 3: Double-digested pVAX1-sFLT-1 with Hind III and ECORI. sFLT-1 was released from double digested recombinant plasmid at the size of interest (1000 bp).
Construction of a recombinant lentiviral vector expressing sFLT-1
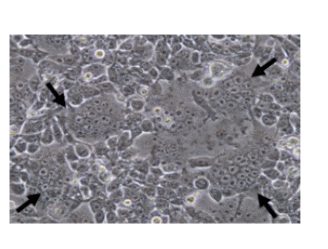
Formation of huge giant cells (syncytium) in 283FT cells cotransfected with pLenti-sFLT-1, and ViraPower™packaging plasmid mix (72 h post- transfection). Arrows indicate huge multinucleated giant cells showing LV-sFLT-1 production in the cells which causes the infected cells fuse together to form large cells with many nuclei.
Verification of expression of sFLT-1 by enzyme-linked immunosorbent assay (ELISA), RT-PCR and western blotting
An optimal amount of LV-sFLT-1 was detected to express the highest level of sFLT-1 protein in transduced cells, and the K562 cells were transduced with different copy numbers of LV-sFLT-1 in 12-well cell culture plates. After 72 h of transduction the supernatants of cells were subjected to sFLT-1 protein quantitation by ELISA. The standard curve was constructed using Excel by plotting the mean absorbance for each standard on the y-axis against the concentration on the x- axis. The concentration of samples was obtained in comparison with the standard curve. 8 × 108 (copies/μl) was chosen as the optimal virus level to transduce the cells. We use this amount of LV-sFLT-1 for all transductions (Figure 4).
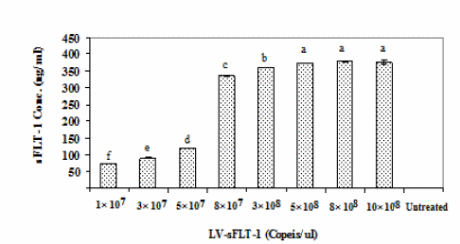
Detection of sFLT-1 protein secretion in K562-sFLT-1 supernatant by ELISA based on the copy numbers of LV-sFLT-1. Different copy numbers of LV-sFLT-1 were used to infect the K562 cells to detect the one which can induce the highest sFLT-1 protein secretion in infected cells. The supernatants of parental K562 cells and K562-sFLT1–cells were taken at 72 h post- infection and subjected to ELISA analysis. The data showed an increased level of sFLT-1 secretion by increasing LV-sFLT-1 copies. A slight difference was detected in the copy numbers 3 × 108, 5× 108, and 8 × 108, while among them the copy number of 8 × 108 showed the highest level of sFLT-1. Therefore this copy number was chosen for transducing cells for further experiments. Each data point is presented as mean ± SEM concentration (ng/ml) of three culture wells.
HUVEC proliferation inhibition assay
The results showed a decrease in HUVEC proliferation compared to the cells treated with the conditioned media of infected cells with LV-EmGFP and the uninfected cells.
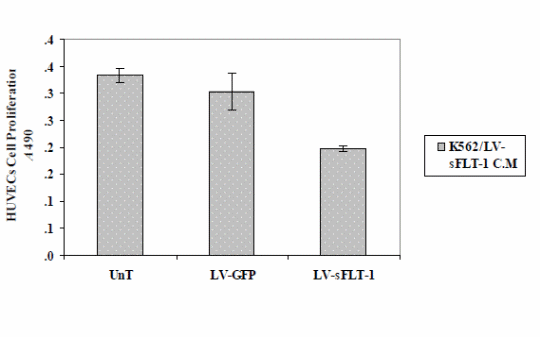
Cell proliferation assay (MTS) to study the effect of sFLT-1 expression from K562 cells transduced with LV-sFLT-1. The conditioned media from K562-sFLT-1, K562-GFP and parental cells were added to HUVECs and incubated in the presence of the VEGF protein 72h post-infection. Cell proliferation was evaluated by MTS assay. The optical density of cells was measured at 490 nm. The results of this study showed a significant decrease in the proliferation of HUVECs when treated with conditioned media from LVsFLT-1 infected K562 cells compared to GFP and untreated controls. Each data point is presented as mean ± SEM. (p<0.05)
HUVEC migration inhibition assay
The findings show that the reduction of cell proliferation was more than that in the non-viral study (p<0.05), while the difference between the two delivery systems on HUVECs migration was not significant (p<0.05) (Figure 6).
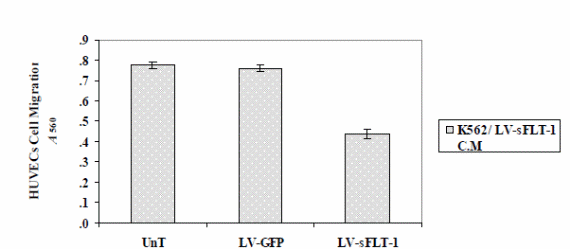
The optical density of migrated cells was measured at 490 nm. The results illustrated that cell migration was markedly reduced in the cells treated with the K562-sFLT-1 conditioned media compared to the GFP and untreated groups. Each data point is presented as mean ± SEM (p<0.05).
Discussion and Conclusion
Approaches aimed at inhibiting the angiogenic process hold a great potential in the management of malignant disorders. An of excess of angiogenic proteins is considered as an essential factor involved in the malignant phenotype in solid tumors as well as hematologic malignancies (Bellamy et al, 1999; Folkman, 1995). VEGF is a potent angiogenic factor involved in angiogenesis- mediated progression of AML. The identification of crucial role of VEGF and VEGFRs in progression of acute myeloid leukemia has led to targeting VEGF and VEGFRs in the antiangiogenic therapeutic approaches for AML. The identification of natural negative regulators f angiogenesis showed that their inhibitory effects are associated to either direct inhibition of the angiogenic process, including endothelial cell proliferation or migration, or, otherwise, indirectly through the suppression of angiogenic factors (such as VEGF) by tumor cells (Schmidt-Wolf & Schmidt-Wolf, 2003). Therefore, these inhibitors are good candidates for gene delivery, because as they produce physiologically, they are not immunogenic, and are also effective extracellularly (Boehm, Folkman, Browder, & O’Reilly, 1997). Thus, gene transfer technology can be a good preference to achieve the expression of such factors in vivo. (Kendall & Thomas, 1993; Kendall, Wang, & Thomas, 1996).
Studies have reported the role of sFlT-1 delivery as an antitumor agent by inhibiting VEGF. Further studies show that VEGF fosters the growth, relocation and survival of tumor cells. Tumor angiogenesis is interplay of the poise of angiogenic and antiangiogenic determinants. Various angiogenic factors have been associated with hematologic malignancies. However, few studies have elaborated the role VEGF (a proangiogenic factor of tumor origin) in such malignancies. Angiogenesis is a multifaceted process and as such, targeting a solitary angiogenic factor may of little impact in total preclusion of angiogenesis (Ye et al, 2004). These include use of naked plasmid DNA and cationic liposome-formulated plasmid DNA. Others such as lentiviral-mediated approaches have been used. The therapeutic effect of Lentivirus-expressed sFLT-1 is occurs via a paracrine system to cause inhibition of angiogenesis. As a result, the therapeutic effect of AdRGDGFPsFLT-1 is independent of the quantitative transduction process. Based on this, the capacity of AdRGDGFPsFLT-1 can be utilized in delivery of an augmented antitumor effect may be of great use in help dodging the confines of vectors to achieve quantitative tumor transduction. As such, we have developed a novel method of counteracting angiogenesis via blockade of VEGF using a specific inhibitor sFLT-1. The use of lentivirus as a delivery system has shown no visible adverse effects in animal models and as such this method provides an important step in gene delivery systems to counter hematologic malignancies.
References
Aguayo, A., Estey, E., Kantarjian, H., Mansouri, T., Gidel, C., Keating, M., et al. (1999). Cellular vascular endothelial growth factor is a predictor of outcome in patients with acute myeloid leukemia. Blood, 94(11), 3717.
Atala, A., & Soker, S. (2002). In vivo administration of vascular endothelial growth factor (VEGF) and its antagonist, soluble neuropilin-1, predicts a role of VEGF in the progression of acute myeloid leukemia in vivo. American Society of Hematology.
Bellamy, W., Richter, L., Frutiger, Y., & Grogan, T. (1999). Expression of vascular endothelial growth factor and its receptors in hematopoietic malignancies. Cancer Research, 59(3), 728.
Boehm, T., Folkman, J., Browder, T., & O’Reilly, M. S. (1997). Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature, 390(6658), 404-407.
Boshart, M., Weber, F., Jahn, G., Dorsch-Hler, K., Fleckenstein, B., & Schaffner, W. (1985). A very strong enhancer is located upstream of an immediate early gene of human cytomegalovirus. Cell, 41(2), 521-530.
Folkman, J. (1971). Tumor angiogenesis: therapeutic implications. New Engl J Med, 285(21), 1182-1186.
Frankel, A. E., & Gill, P. S. (2004). VEGF and myeloid leukemias. Leukemia Research, 28(7), 675-677.
Grimwade, D. (2001). The clinical significance of cytogenetic abnormalities in acute myeloid leukaemia. Best Practice & Research Clinical Haematology, 14(3), 497-529.
Kamps, W., De Bont, E., Fidler, V., Meeuwsen, T., Scherpen, F., &Hählen, K. (2002). Vascular endothelial growth factor secretion is an independent prognostic factor for relapse-free survival in pediatric acute myeloid leukemia patients. Clinical cancer research, 8(9), 2856.
Kendall, R. L., & Thomas, K. A. (1993). Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proceedings of the National Academy of Sciences of the United States of America, 90(22), 10705.
Kendall, R. L., Wang, G., & Thomas, K. A. (1996). Identification of a natural soluble form of the vascular endothelial growth factor receptor, sFLT-1, and its heterodimerization with KDR* 1. Biochemical and Biophysical Research Communications, 226(2), 324-328.
Schuch, G., Machluf, M., Bartsch Jr, G., Nomi, M., Richard, H., Atala, A., et al. (2002). In vivo administration of vascular endothelial growth factor (VEGF) and its antagonist, soluble neuropilin-1, predicts a role of VEGF in the progression of acute myeloid leukemia in vivo. Blood, 100(13), 4622.
Hussong, J., Rodgers, G., & Shami, P. (2000). Evidence of increased angiogenesis in patients with acute myeloid leukemia. Blood, 95(1), 309.
Kelly, L., & Gilliland, D. (2002). Genetics of myeloid leukemias. Annual review of genomics and human genetics, 3(1), 179-198.
Kerbel, R. (2000). Tumor angiogenesis: past, present and the near future. Carcinogenesis, 21(3), 505.
Kume, A., Hanazono, Y., Mizukami, H., Okada, T., & Ozawaa, K. (2002). Selective expansion of transduced cells for hematopoietic stem cell gene therapy. International journal of hematology, 76(4), 299-304.
List, A. (1999). Pharmacological differentiation and anti-apoptotic therapy in myelodysplastic syndromes.
Padro, T., Ruiz, S., Bieker, R., Burger, H., Steins, M., Kienast, J., et al. (2000). Increased angiogenesis in the bone marrow of patients with acute myeloid leukemia. Blood, 95(8), 2637.
Ribatti, D., Vacca, A., & Presta, M. (2000). The discovery of angiogenic factors: A historical review. General Pharmacology: The Vascular System, 35(5), 227-231.
Schmidt-Wolf, G., & Schmidt-Wolf, I. (2003). Gene therapy for hematological malignancies. Clinical and experimental medicine, 3(1), 4-14.
Ye C, Feng C, Wang S, Wang KZ, Huang N, Liu X, Lin Y et al. (2004). sFLT-1 gene therapy of follicular thyroid carcinoma. Endocrinology 145: 817–822